Interaction energies
In this tutorial, we will explore the calculation of molecular interaction energies using the InteractionEnergy workflow.
Specifically, we will focus on studying the interaction energy of a water dimer.
By studying the interaction energy, we gain insight into the strength and nature of interactions between molecules.
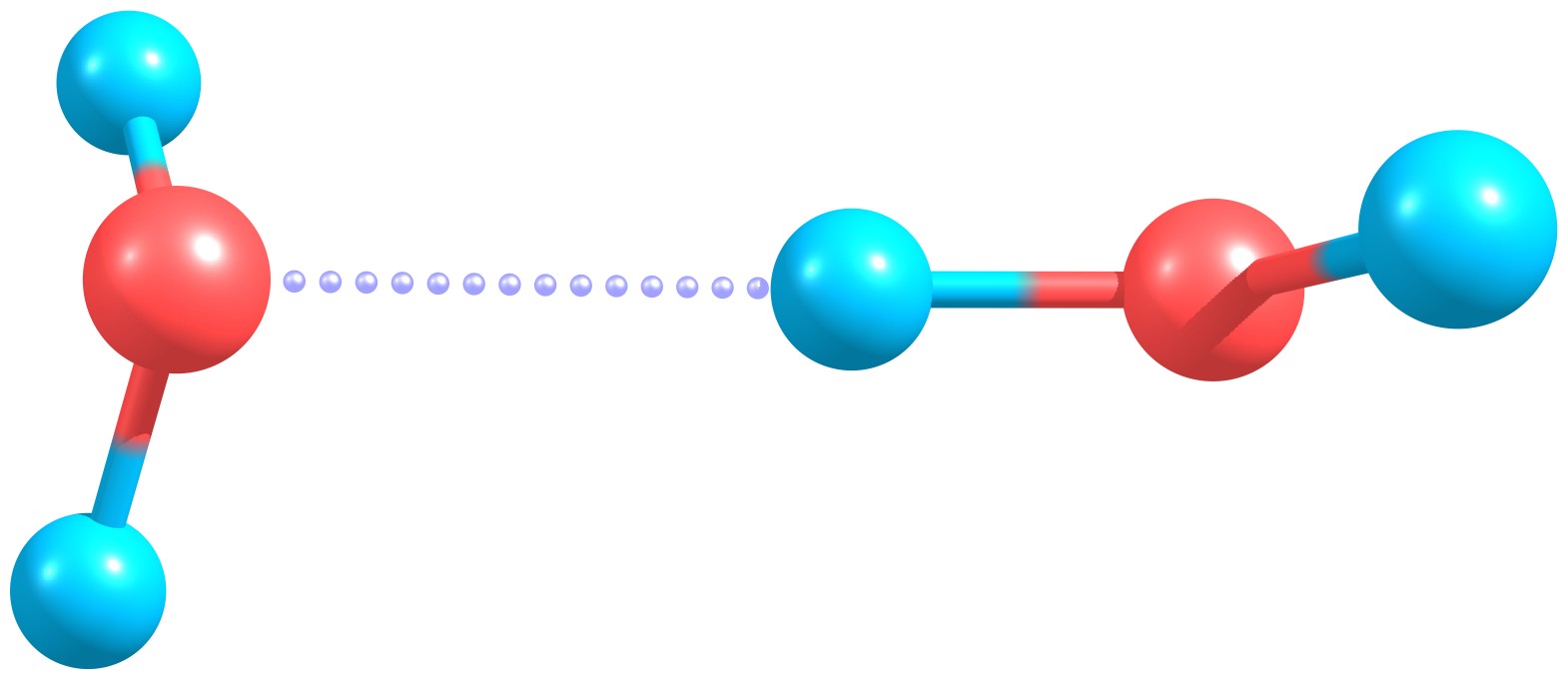
Water dimer.
How to run the calculation
To perform the calculation yourself,
you can download the XYZ file waterDimer.xyz containing the structure of the water dimer.
Note
WEASEL recognizes the monomers of the complex by detecting the bonds based on simple GFN2-xTB calculations.
Therefore, it is only necessary to provide the structure of the dimer.
If the structure is provided via a .mol2 file, the bond information in the .mol2 file is used.
Important
The molecular interaction workflow works only for complexes with two monomers.
If you are studying complexes consisting of more than two monomers, you can still run the
molecular interaction workflow by providing a .mol2 file in which you assign bonds between those molecules,
that should be treated as one monomer in the calculation.
Once you have the file ready, you can start the molecular interaction workflow by running the following command in your terminal:
weasel waterDimer.xyz -W InteractionEnergy
Steps of the WEASEL workflow
The interaction energy workflow consists of several key steps:
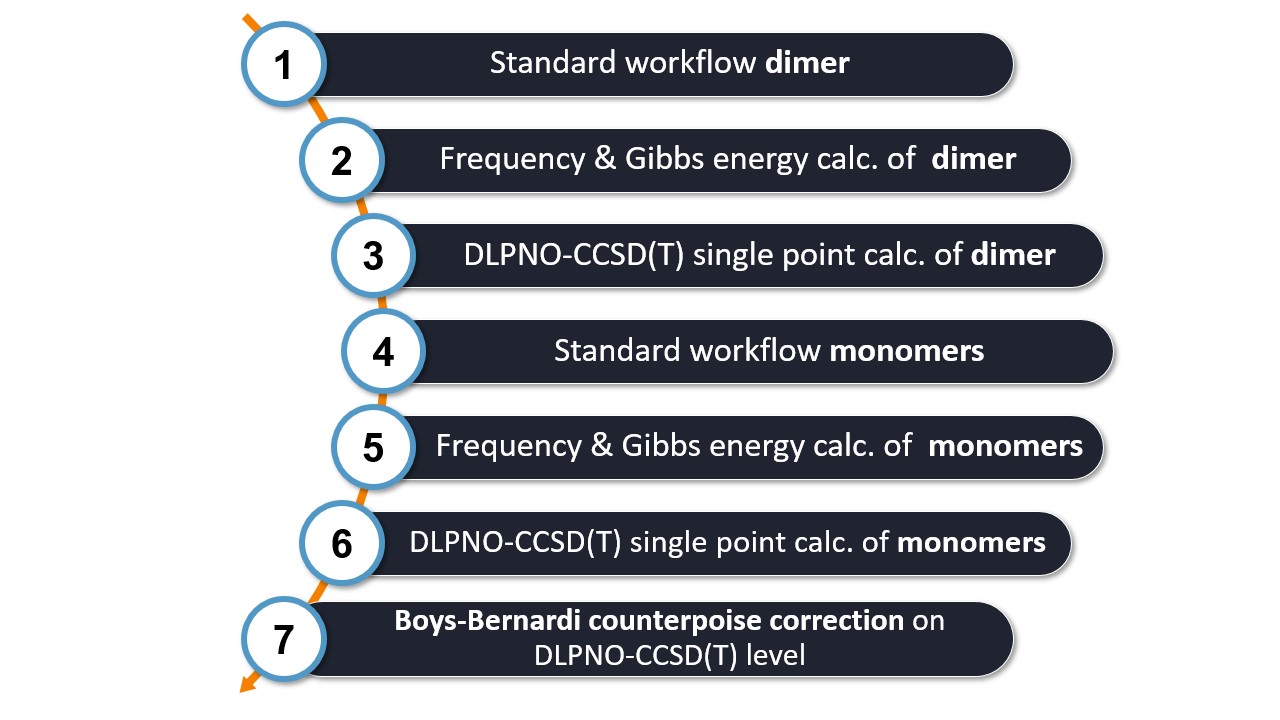
For the calculations, WEASEL utilises ORCA as the backend QC driver. Let's look at each step in more detail:
Step 1: Inside WEASEL, a pre-optimization, optimization, and DFT single point energy calculations on the dimer system are conducted.
Step 2: The frequencies and Gibbs energies of the dimer are calculated.
Step 3: A DLPNO-CCSD(T) single point energy calculation on the dimer is conducted. This high level calculation gives us accurate energy values for the dimer system.
Note
You can modify the default methods used for workflow steps 1-3 according to your needs and preferences using the provided keyword arguments.
Step 4, 5, 6: WEASEL repeats steps 1 to 3 for each of the monomers involved in the interaction. This ensures that we have accurate energy values for each monomer.
Step 7: Finally, WEASEL applies the Boys-Bernardi counterpoise correction to the DLPNO-CCSD(T) energy calculations of the monomers. This correction accounts for the basis set superposition error (BSSE) and provides a more accurate estimation of the interaction energy.
Explanation
The BSSE correction addresses errors that occur when two molecules are close together and share the same basis set, leading to an overestimation of the interaction energy. The Boys-Bernardi Counterpoise Correction is a type of BSSE correction that accounts for this error by adding an additional basis set to the monomer calculations.
With these steps, WEASEL provides reliable and comprehensive results for the interaction energy between molecules.
The progress of each step during the calculation can be tracked in the .report file. In the next section you will see that WEASEL automatically summarizes each individual energy as well as the total interaction energies, which are easily accessible in the .summary file.
Output files and results
After running the workflow, you will find several files generated in the main job directory waterDimer_InteractionEnergy. The directory tree is shown below:
.
├── waterDimer.xyz
└── waterDimer_InteractionEnergy
├── waterDimer_InteractionEnergy.input.xyz
├── waterDimer_InteractionEnergy.report
├── waterDimer_InteractionEnergy.results.xyz
├── waterDimer_InteractionEnergy.summary
├── waterDimer_Opt.xyz
├── BuildTopo
│ └── waterDimer_BuildTopo job files
├── PreOpt
│ └── waterDimer_PreOpt job files
├── Opt
│ └── waterDimer_Opt job files
├── Freq
│ └── waterDimer_Freq job files
├── SP_WF
│ └── waterDimer_SP_WF job files
└── bsse-BB
└── waterDimer_bsse-BB job files
Inside the waterDimer_InteractionEnergy directory you will find several subdirectories corresponding to different calculation steps such as Setup, PreOpt, Opt, Freq, SP_WF, and bsse-BB.
Note
The files related to the monomers (PreOpt, Opt, SP_WF) can be found in the subdirectory bsse-BB.
You can see from the directory tree that within the mainjob directory itself there are only files known from the basic workflow of WEASEL.
No files specific to the interaction energy workflow are created.
The important information about the interaction energy is already stored in the standard files - the .report and the .summary file.
The file waterDimer_InteractionEnergy.summary contains the data of all calculation steps in the order they were performed.
We will focus on the last few entries in this file, which provide crucial information about the interaction energy.
These include the Boys-Bernardi counterpoise correction, the interaction energy without the counterpoise correction,
the interaction energy with the counterpoise correction, and the interaction energy with the additional thermochemical correction.
Energy [kcal/mol] / Value Type Calculation type Method Basis set Solvent Charge Multiplicity Further tags
-0.821129 CP-correction bsse-BB DLPNO-CCSD(T) def2-TZVPP Gas phase 0 1 bsse-BB Summary WF
-3.309265 SP-Energy Interaction-Energy DLPNO-CCSD(T) def2-TZVPP CPCM(Water) 0 1 bsse-BB Summary WF
-2.488136 CP-corrected SP-Energy Interaction-Energy DLPNO-CCSD(T) def2-TZVPP CPCM(Water) 0 1 bsse-BB Summary WF
5.342636 CP-corrected Gibbs Energy G WF Interaction-Energy DLPNO-CCSD(T) def2-TZVPP CPCM(Water) 0 1 bsse-BB Summary WF
In addition, a summary of the interaction energies is provided at the end of the .report file,
highlighting the values for the Boys-Bernardi counterpoise correction,
the interaction energy without counterpoise correction,
the interaction energy with counterpoise correction, and the interaction free energy with counterpoise correction:
*****************************************
BSSE: SUMMARY WF (Type: bsse-BB)
*****************************************
Adding CP correction -0.821129 kcal/mol to summary file.
Adding interaction energy (no CP correction) -3.309265 kcal/mol to summary file.
Adding interaction energy (with CP correction) -2.488136 kcal/mol to summary file.
Adding interaction free energy (with CP correction) -3.309265 kcal/mol to summary file.
#################################### ACTIONS CONCLUDED #####################################
Looking at these files, you can see that the interaction energy of the two water molecules is about -3.3 kcal/mol when not only the counterpoise correction but also the free energy is considered.