Optimal Binding Workflow
In this tutorial we will explore the optimal binding workflow in WEASEL, which is specifically designed to identify optimal conformer ensembles in metal-ligand complexes.
The optimal binding workflow begins by using the docker, as shown in the tutorial found here, to effectively generate the metal-ligand complex. However, it is recognized that this initial assembly may not always result in the lowest energy conformer. To address this limitation, and to account for scenarios where multiple conformers play an important role in the metal-ligand complex, the workflow includes a conformer search after the initial Docker assembly.
The optimal binding workflow differs from the standard conformer search workflow, which can be found in the tutorial here, in that the optimal binding workflow addresses potential shortcomings of the standard conformer search workflow that may arise when dealing with a metal-ligand complex assembled from scratch.
The optimal binding workflow introduces two major changes from the standard conformer search. First, it uses GOAT (Global Optimization Approach Tool) in conjunction with a specific method within GOAT to curate the most diverse conformers for the initial conformer set. This approach ensures a comprehensive exploration of potential complexes and increases the accuracy of the results.
Second, the standard conformer search relies on XTB levels for pre-optimization of the initial conformer ensemble. However, XTB may not be the best fit for capturing non-covalent interactions that are prevalent in metal-ligand complexes. To address this issue, the XTB pre-optimization step is replaced with a DFT pre-optimization using loose optimization criteria. This strategic change avoids problems such as over-coordination of metal centers that can often occur with XTB.
In this tutorial, we will explore the optimal binding workflow using the manganese complex [Mn(CDTA)(H2O)]2- as an example.
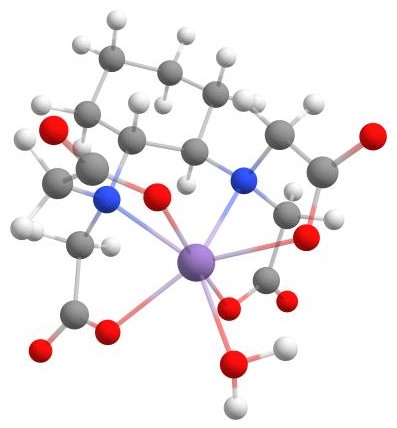
The example [Mn(CDTA)(H2O)]2-.
How to run the calculation
If you want to follow along with the tutorial, you can download the XYZ coordinates provided below to assemble the metal-ligand complex:
If you create the XYZ files yourself, make sure that the comment line (second line in the XYZ file) contains the charge and multiplicity of the systems: for CDTA -2 and 1, for water 0 and 1, and for manganese 2 and 6. This is relevant for the Docker tool because it reads the charge and multiplicity of each added system directly from the file, as described in the Docker tutorial.
To assemble the complex and use the optimal binding workflow to find the metal-ligand complex, use the following command:
$ weasel mn.xyz -c 2 -m 6 -dock cdta.xyz h2o.xyz -W OptimalBinding
You can alternatively specify the manganese ion directly in the calculation using the keyword -atom
followed by the atom type, here Mn, and its charge, here 2, and multiplicity, here 6.
$ weasel -atom mn -c 2 -m 6 -dock cdta.xyz h2o.xyz -W OptimalBinding
The -dock cdta.xyz h2o.xyz part of the command adds the CDTA ligand to the manganese first, followed by the water ligand.
Then -W OptimalBinding initiates the search for the optimal metal-ligand complex conformer or conformer ensemble.
Steps of the WEASEL workflow
The optimal binding workflow automatically applies a series of steps to the assembled structure generated by the Docker. The figure below provides an overview of the steps in the workflow.

Let's take a closer look at each step:
1. Generate the Conformer Ensemble: First, the Global Optimization Approach Tool (GOAT), along with a specific setting, generates a geometrically diverse conformer ensemble. This process allows a wide variety of geometrically diverse conformers to be explored, rather than just focusing on the lowest energy conformers. GOAT considers the fragments defined by the Docker, which in this case include manganese, CDTA, and water.
2. Initial Conformer Filtering: Next, an initial filtering step is performed to eliminate any duplicate conformers in the ensemble.
3. Loose Optimization Step: A pre-optimization step is performed using loose optimization criteria with the rScan-3c DFT function. Energy filtering is applied to retain only the lowest energy conformers with an energy range less than 6 kcal/mol.
4. Conformer Ensemble Optimization: The conformer ensemble is optimized to further refine the structures. An energy filter of 5.00 kcal/mol is applied to ensure that only the most stable conformers are considered in the next step.
5. Single Point DFT Calculation: A single point DFT calculation is performed using the wB97X-V functional with a def2-TZVP basis set. This step provides a higher level of accuracy for the energy calculations. An energy filter of 4.00 kcal/mol is used in this final filtering step to provide the most reliable and accurate results.
6. Frequency calculation of the lowest conformer: Since the thermodynamic state is important for the stability of a complex, in this step the frequencies of the lowest identified conformer are calculated to determine its Gibbs free energy. This is calculated only for the lowest conformer as the goal of the workflow is to identify the most stable complex.
7. Ensemble thermodynamic state function calculation: An additional Gibbs free energy correction can be achieved by considering the influence of the conformer ensemble. Therefore in this last step the ensemble thermodynamic state functions are determined via GOAT together with XTB. If you do not want to calculate this additional correction, you can switch it off by adding the keyword
-no-ensemblethermo.
By following these steps, the Optimal Binding workflow allows us to identify the most appropriate metal-ligand complex conformer or conformer ensemble with a comprehensive and accurate approach.
Output files and results
The optimal binding workflow generates several files that provide information about the calculation and results:
.
├── mn.xyz
└── mn_OptimalBinding
├── mn_Docking.docker.xyz
├── mn_OptimalBinding.report
├── mn_OptimalBinding.results.xyz
├── mn_OptimalBinding.summary
├── Docking
│ └── mn_Docking job files
├── ConfSearch
│ └── mn_ConfSearch job files
The most important files and a brief description of their contents are listed in the table below.
File |
Description |
|---|---|
Cobalt.results.xyz |
Final optimized host-guest structure after WEASEL optimal
binding workflow
|
Cobalt_Docking.docker.xyz |
Host-guest structure after docking |
Cobalt.report |
Information about each step of the calculation and distances between
host and guest in final conformer
|
Cobalt.summary |
Summary of results including XTB level host-guest interaction energies
after docking and energies of all conformers after each filtering step
including final energy of lowest-energy conformer
|
Please refer to the Docker tutorial for information on output files specific to the docking process. After the specific results are written to the summary file related to the docker, the summary file follows the same structure as explained in the tutorial about conformational searches.
Note that Cobalt_Docking.docker.xyz contains the structure after the docking process,
whereas Cobalt.results.xyz contains the final result of the conformer search.
The figure below compares the structure of the assembled structure and the lowest-energy conformer.
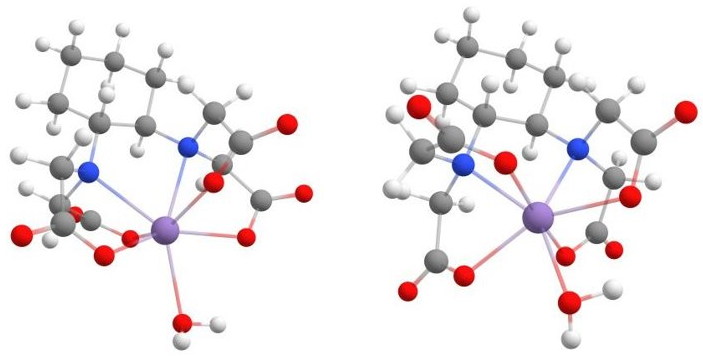
Sturcture of [Mn(CDTA)(H2O)]2- resulting from the Docker (left) and from the optimal binding workflow (right).
Keywords and remarks
Keyword |
Description |
|---|---|
|
Ignore H-atoms for conformer screening. |
|
Include H-atoms for conformer screening. |
|
Set GOAT-DIVERSITY option to focus on geometrical diversity. |
|
Free topology between different fragments. |
|
For conformer searcher with GOAT, constrain maximum
coordination number (CN) for individual atoms.
Two syntaxes are allowed to specify the max CN:
1) ID:CN, e.g. 3:4, where ID is the position
of the atom in the input file and CN is the coordination
number; 2) ELEMENT:CN, e.g. Mn:5, where all atoms of
type ELEMENT will have a maximum CN of 5.
To specify multiple entries, separate them by spaces:
e.g. 5:3 C:4 ...
|