Scans
In this tutorial we will explore the concept of relaxed surface scans, which is of great interest in computational chemistry. Relaxed surface scans involve systematically varying molecular geometries to analyze the potential energy surface, for example to study the energetics of molecules or to find different stable structures of a system. These scans can be performed along different degrees of freedom, such as bond lengths, angles, and dihedrals. In this tutorial we will specifically focus on performing a dihedral scan with WEASEL to study the rotation of ethane around its axis, providing insight into its energy landscape and its rotational barrier. The following figure visualized the example.
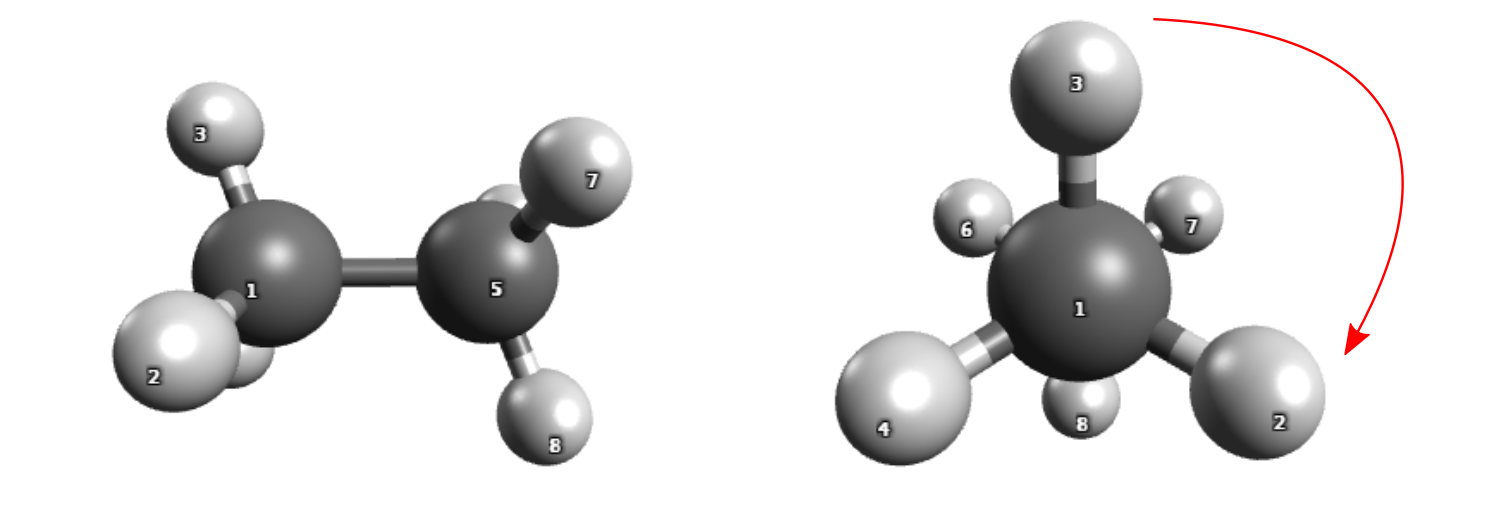
Rotation around the central bond of ethane.
At the end of the tutorial there is also a list of keywords to study the other degrees of freedom with WEASEL's scan function.
How to run the calculation
If you want to carry out the calculation yourself, you can download the file XYZ coordinates
file ethane.xyz.
To rotate the CH 3 groups around each other, you need to specify the atomic indices (see figure above for indices) that define the torsion angle to be rotated. Follow these steps:
1. Use the command -scan-dihedral 3 1 5 7 to specify the atoms H3, C1, C5, and H7 involved in the rotation,
which will rotate the two CH 3 units around the C1-C5 bond.
Set the start angle to 60° with
-scan-start 60and the stop angle to 180° with-scan-stop 180.For a more accurate energy surface, perform a wave function calculation by adding the
-spwfoption.
The complete command to run in WEASEL is:
weasel ethane.xyz -scan-dihedral 3 1 5 7 -scan-start 60 -scan-stop 180 -spwf
Warning
If you are already used to the ORCA input format, note that WEASEL starts counting atom indices at 1 by default. This can be changed in the default settings:
[SYSTEM]
StartIndexingAtoms = 0
Steps of the WEASEL workflow
The figure below summarizes the steps WEASEL performs after the above command is requested:
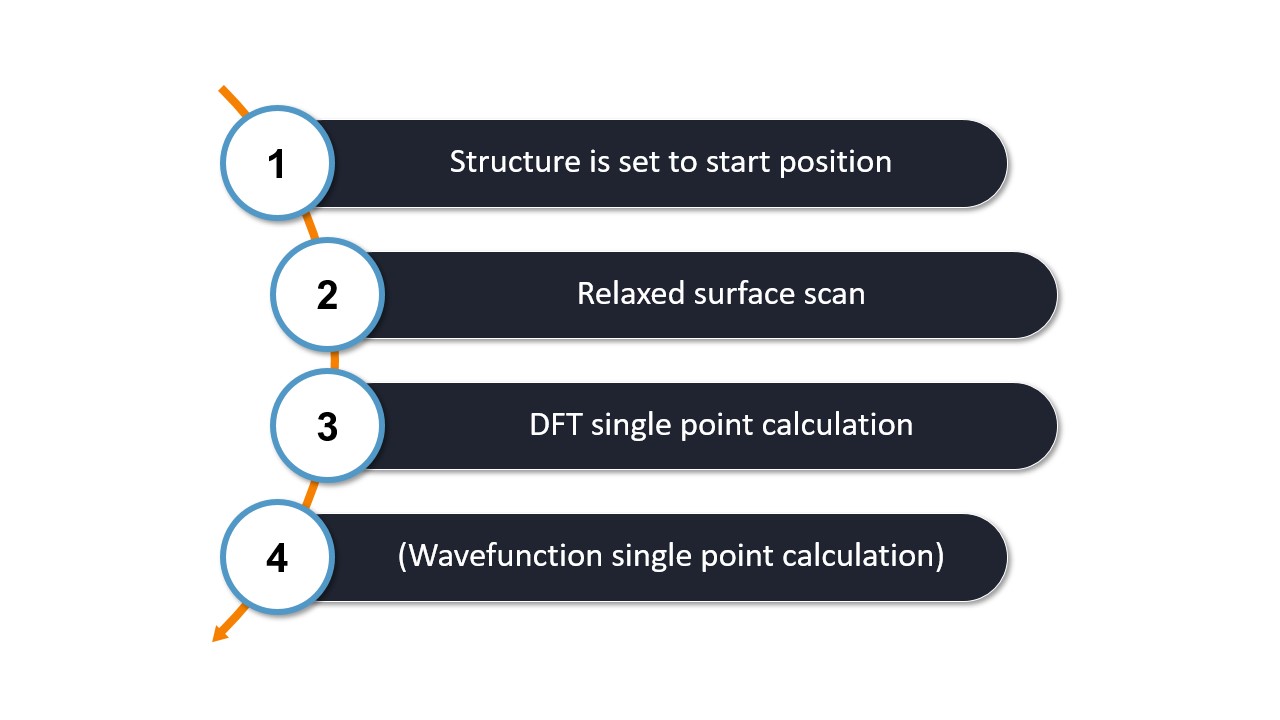
Step 1: The simulation begins by rotating the ethane molecule to the scan start angle.
Step 2: A relaxed surface scan is performed along the dihedral angle using ORCA.
The relaxed surface scan performs ten steps by default. You can change the number of steps by adding the keyword -scan-nsteps followed by the desired number of steps.
Step 3 and Step 4: For each of the ten structures resulting from the relaxed surface, a single-point DFT calculation is performed first, followed by a single-point wavefunction calculation.
Output files and results
The calculation produces the following files:
.
├── ethane.xyz
└── ethane
├── ethane_PreOpt.xyz
├── ethane_Scan.allxyz
├── ethane_Scan.xyz
├── ethane.report
├── ethane.summary
├── Opt
│ └── ORCA job files
├── PreOpt
│ └── ORCA job files
└── SP_DFT
└── ORCA job files
The energies of the different structures are stored in the summary file:
Summary file for job: weasel ethane.xyz -scan-dihedral 3 1 5 7 -scan-start 60 -scan-stop 180 -finalWF
-50111.379351 SP-Energy Scan RI-BP86 def2-TZVP(-F) Gas phase 0 1 Scan-Step(D 3 1 5 7 = 60.000)
-50111.101586 SP-Energy Scan RI-BP86 def2-TZVP(-F) Gas phase 0 1 Scan-Step(D 3 1 5 7 = 73.333)
...
-50111.379722 SP-Energy Scan RI-BP86 def2-TZVP(-F) Gas phase 0 1 Scan-Step(D 3 1 5 7 = 180.000)
-50077.846718 SP-Energy SP_DFT RIJCOSX-B3LYP def2-TZVP Gas phase 0 1 Scan-Step(D 3 1 5 7 = 60.000)
-50077.555975 SP-Energy SP_DFT RIJCOSX-B3LYP def2-TZVP Gas phase 0 1 Scan-Step(D 3 1 5 7 = 73.333)
...
-50077.846473 SP-Energy SP_DFT RIJCOSX-B3LYP def2-TZVP Gas phase 0 1 Scan-Step(D 3 1 5 7 = 180.000)
-49986.627431 SP-Energy SP_WF DLPNO-CCSD(T) def2-TZVP Gas phase 0 1 Scan-Step(D 3 1 5 7 = 60.000)
-49986.329479 SP-Energy SP_WF DLPNO-CCSD(T) def2-TZVP Gas phase 0 1 Scan-Step(D 3 1 5 7 = 73.333)
...
-49986.627181 SP-Energy SP_WF DLPNO-CCSD(T) def2-TZVP Gas phase 0 1 Scan-Step(D 3 1 5 7 = 180.000)
The relative energies for both the DFT and wavefunction calculation can be plotted against the torsion angle:
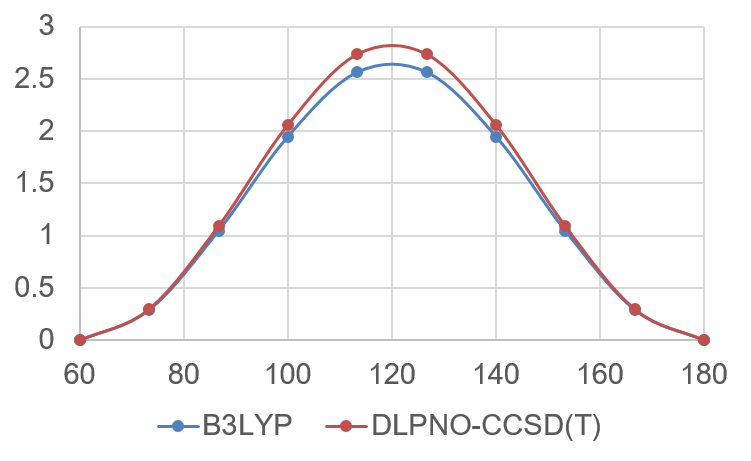
Energy plot from the relaxed surface scan around the ethane central torsion angle.
This plot provides an approximation to the rotational barrier of ethane around the central bond. The value can be compared to the value that is obtained with a proper TS optimization in the reactivity workflow.
Remarks and keywords
-start and -stop values of the scan are required. WEASEL can carry out scans on bond distances, bond angles, and torsion angles.
Keyword |
Description |
|---|---|
|
Value at which the scan starts. [Angstrom] for bond scans, [degrees]
for angle and torsion scans.
|
|
Value at which the scan starts. [Angstrom] for bond scans, [degrees] for angle
and torsion scans.
|
|
Number of steps for the scan. Default value is 10, defined in the
OPT section of the default settings.
|
|
Scan the bond distance between atom
INT2 and INT2.Should be accompanied by the
-scan-start and -scan-stop keyword.Distances in [Angstrom].
|
|
Scan the bond angle between atom
INT1, INT2 and INT3.Should be accompanied by the
-scan-start and -scan-stop keyword.Angles in [degrees].
|
|
Scan the dihedral angle between atom
INT1, INT2, INT3 and INT4.Should be accompanied by the
-scan-start and -scan-stop keyword.Angles in [degrees].
|
Note
Input coordinates can be different to the scan-start value.