Reactivity workflow
The tendency of substances to undergo a chemical reaction is determined by the activation and reaction energies of the reaction. The activation energy is the energy difference between the reactants and the transition state, while the reaction energy is the energy difference between the reactants and the products. To conveniently study reactions, you can use the Reactivity Workflow, which automatically calculates the energies of the reactants, transition states, and products, and the corresponding activation and reaction energies. The workflow uses the Nudged Elastic Band (NEB) technique, which optimizes intermediate structures along a path connecting the reactants and products of a chemical reaction. NEB calculations are commonly used to determine the activation energies and transition states of chemical reactions.
In the following, some examples are given where the Reactivity Workflow has been used. The basics of the workflow are explained using the example of the rotational barrier of ethane, followed by the Diels-Alder mechanism and the study of rotational groups.
Rotational barrier
We have a look at the rotational barrier of ethane, from the staggered configuration via the eclipsed to the staggered configuration.
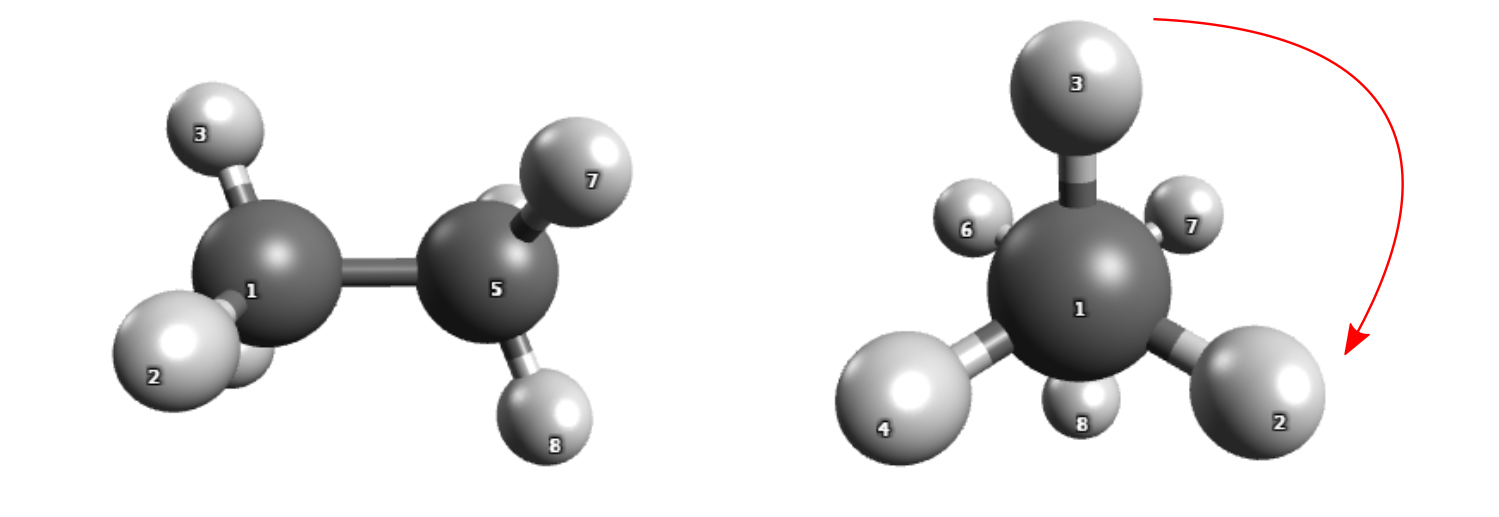
Rotation around the central bond of ethane.
How to run the calculation
In principle, the xyz coordinates of ethane introduced in Scans can be used. However, the reactivity
workflow requires both reactant as well as product structure as input.
The XYZ files of the reactant (ethane_reac.xyz) and the
product (ethane_prod.xyz)
ethane can be downloaded to the directory in which you want to run the calculation.
Important
In case you create the XYZ files yourself: In order for the NEB-TS to work, the atom numbering in both reactant and product geometries must be the same. Which means that a given carbon on the first, should have the same order on the xyz table in the last. The best way to guarantee this is to start from one geometry and move the atoms to draw the other.
The workflow can be run with the following command, specifying the product structure with the -product keyword.
weasel ethane_reac.xyz -W Reactivity -product ethane_prod.xyz
Steps of the reactivity workflow
The workflow optimizes the reactant and the product geometry and it finds the TS that connects both.
The following calculation steps are the basis of the workflow.
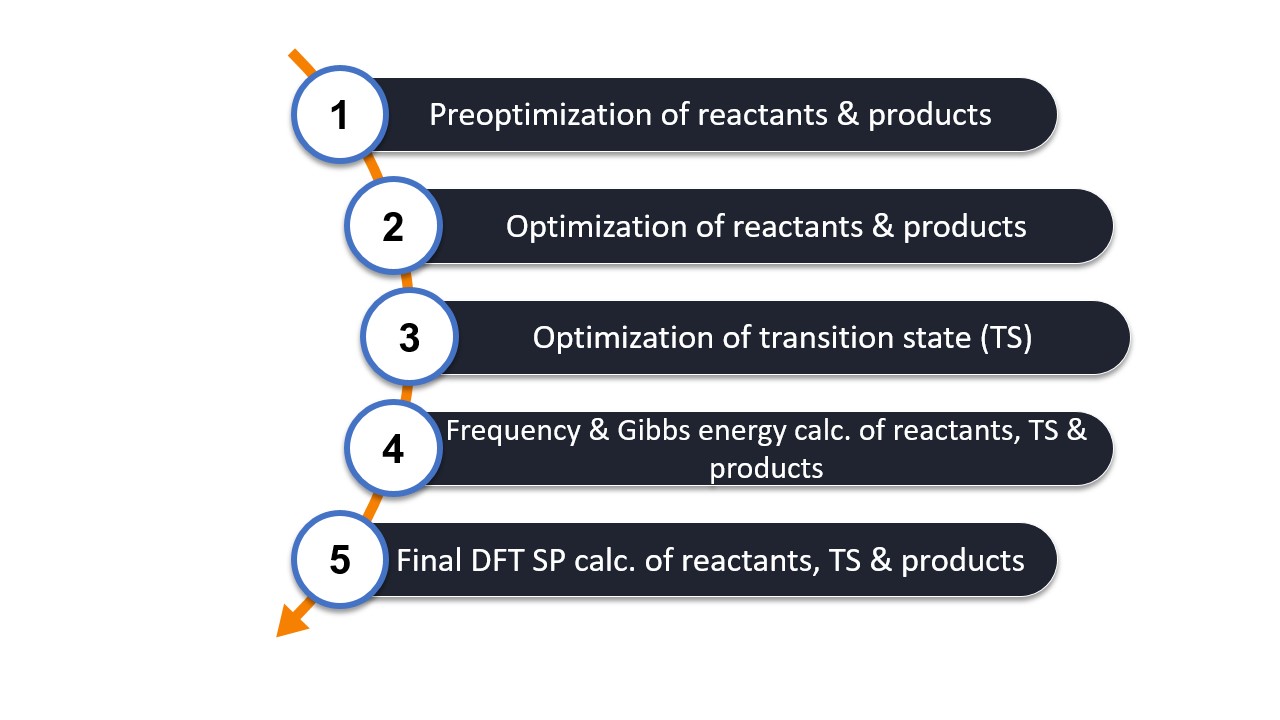
The frequency calculation is required to
check for the (minimum and TS) nature of the optimized structures and
the thermochemical correction using the RRHO approximation.
The final SP energy calculation recomputes all three structures at a more accurate DFT level, here based on the basic workflow with wB97X-V/def2-TZVP.
Recommendation
Highly accurate activation and reaction free energies can be critical for reactivity calculations.
To obtain a more accurate result, it is recommended to recalculate the final SP energy with a SP wavefunction based method,
e.g. DLPNO-CCSD(T) by adding the keyword -spwf.
Note
In addition to the Reactivity workflow, there is also the Reactivity-Explore workflow. They differ in that
Reactivity uses the standard methods for pre-optimization (XTB), optimization and frequency calculation (r2Scan-3c),
while Reactivity-Explore uses XTB by default for all three steps. This makes the latter faster but less accurate.
The methods can also be changed via the respective keyword input.
Note
The solvent in the Reactivity standard workflow is water and needs to be adapted if necessary.
Structure of output files
The calculation produces the following files:
.
├── ethane_reac.xyz
├── ethane_prod.xyz
└── ethane
ethane_reac_ethane_prod_exploreReactivity
├── ethane_reac_ethane_prod_Reactivity.input.xyz
├── ethane_reac_ethane_prod_Reactivity_MEP_trj.xyz
├── ethane_reac_ethane_prod_Reactivity_prod_Opt.xyz
├── ethane_reac_ethane_prod_Reactivity_Product.input.xyz
├── ethane_reac_ethane_prod_Reactivity_reac_Opt.xyz
├── ethane_reac_ethane_prod_Reactivity.report
├── ethane_reac_ethane_prod_Reactivity.restart.tsv.bz2
├── ethane_reac_ethane_prod_Reactivity.summary
├── ethane_reac_ethane_prod_Reactivity_TS_Opt_Freq.hess.v006.xyz
├── ethane_reac_ethane_prod_Reactivity_TS_Opt.xyz
├── BuildTopo
│ └── ORCA job files
├── PreOpt
│ └── ORCA job files
├── Opt
│ └── ORCA job files
├── NEB
│ └── ORCA job files
├── SP_DFT
└── ORCA job files
Below is a table that outlines the most important files created and their respective descriptions.
File |
Description |
|---|---|
stem_reac_Opt.xyz |
optimized reactant structure |
stem_prod_Opt.xyz |
optimized product structure |
stem_TS_Opt.xyz |
optimized TS structure |
stem_TS_Opt_Freq.hess.v006.xyz |
Trajectory file with the TS mode. Can be visualized with avogadro |
stem_NEBTS_MEP_trj.xyz |
Minimum energy path of the converged NEB calculation |
stem_exploreReactivity.report |
Output file of the WEASEL run |
stem_exploreReactivity.summary |
Summary file for the WEASEL run |
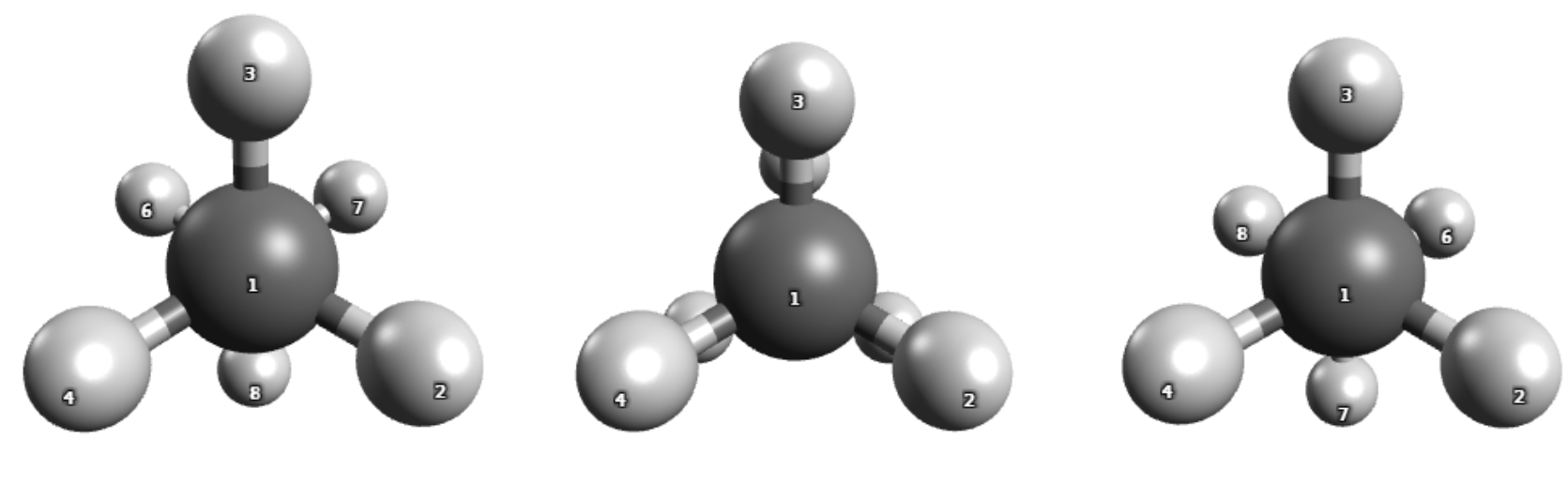
Reactant (left), TS (middle) and product (right) structure.
Note
Further information on the minimum-energy path is found in the NEB task directory.
Reaction and activation free energies
The summary file stores different sets of data, but we'll focus on the most important ones here. At its very end, the activation and reaction energy and free energy at DFT level:
-0.000233 Reaction Energy dE TD RIJCOSX-B3LYP def2-TZVP(-F) CPCM(Water) 0 1 NEB-TS Summary DFT
2.571387 Activation Energy dE TS RIJCOSX-B3LYP def2-TZVP(-F) CPCM(Water) 0 1 NEB-TS Summary DFT
-0.007393 Reaction Free Energy dG TD RIJCOSX-B3LYP def2-TZVP(-F) CPCM(Water) 0 1 NEB-TS Summary DFT
2.537263 Activation Free Energy dG TS RIJCOSX-B3LYP def2-TZVP(-F) CPCM(Water) 0 1 NEB-TS Summary DFT
as well as at wavefunction level:
0.000257 Reaction Energy dE TD DLPNO-CCSD(T) def2-TZVP CPCM(Water) 0 1 NEB-TS Summary WF
2.818516 Activation Energy dE TS DLPNO-CCSD(T) def2-TZVP CPCM(Water) 0 1 NEB-TS Summary WF
-0.006903 Reaction Free Energy dG TD DLPNO-CCSD(T) def2-TZVP CPCM(Water) 0 1 NEB-TS Summary WF
2.784392 Activation Free Energy dG TS DLPNO-CCSD(T) def2-TZVP CPCM(Water) 0 1 NEB-TS Summary WF
are provided as key results of the simulation.
Diels-Alder reaction
More interesting is the formation of new bonds, as in the following example of the Diels-Alder reaction of cyclopentadiene.
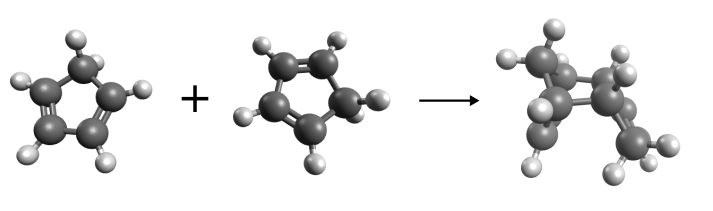
Diels-Alder reaction of cyclopentadiene.
In the following, we will compare the Diels-Alder reaction between the two cyclopentadienes with a reaction between a cyclopentadiene and a methylcyclopentadiene. The purpose of this comparison is to demonstrate that the reactivity workflow in WEASEL can be used for reactions involving bond formation, and to illustrate the potential for transferring results from one WEASEL calculation to another, thereby streamlining and accelerating the process. Let's start with the example reaction between the two cyclopentadienes.
Reaction between two cyclopentadienes
If you want to do the calculation between the two cyclopentadienes yourself,
you can download the XYZ file DA_reac.xyz of the reactant complex
and the file DA_prod.xyz for the product
and save them in the directory where you want to do the calculation.
If you create the structures yourself,
make sure that the atoms have the same order in both the reactant and product structures.
The file DA_reac.xyz already contains the two molecules that will react, positioned close together to form the reactant complex.
Instead of manually searching for a suitable reactant complex with multiple interacting molecules, as in this Diels-Alder example,
you can use the Docker in WEASEL to generate the required complex. The Docker tutorial can be found here.
To perform the calculation, you can use the reactivity workflow, as already applied to the rotational barrier of ethane, by running the following command:
weasel DA_reac.xyz -W Reactivity -product DA_prod.xyz
Recommendation
In this scenario, the reactant complex has many degrees of freedom.
Consequently, during structure optimization,
there is a chance that points on the potential energy surface with multiple negative frequencies
will be mistaken for a local minimum. To reduce the likelihood of this happening in your calculation,
you can use the -correctimagmodes keyword. This will automatically repeat the optimization if any (unwanted)
imaginary modes are detected during the frequency analysis.
The calculation produces the minimum energy path shown below:
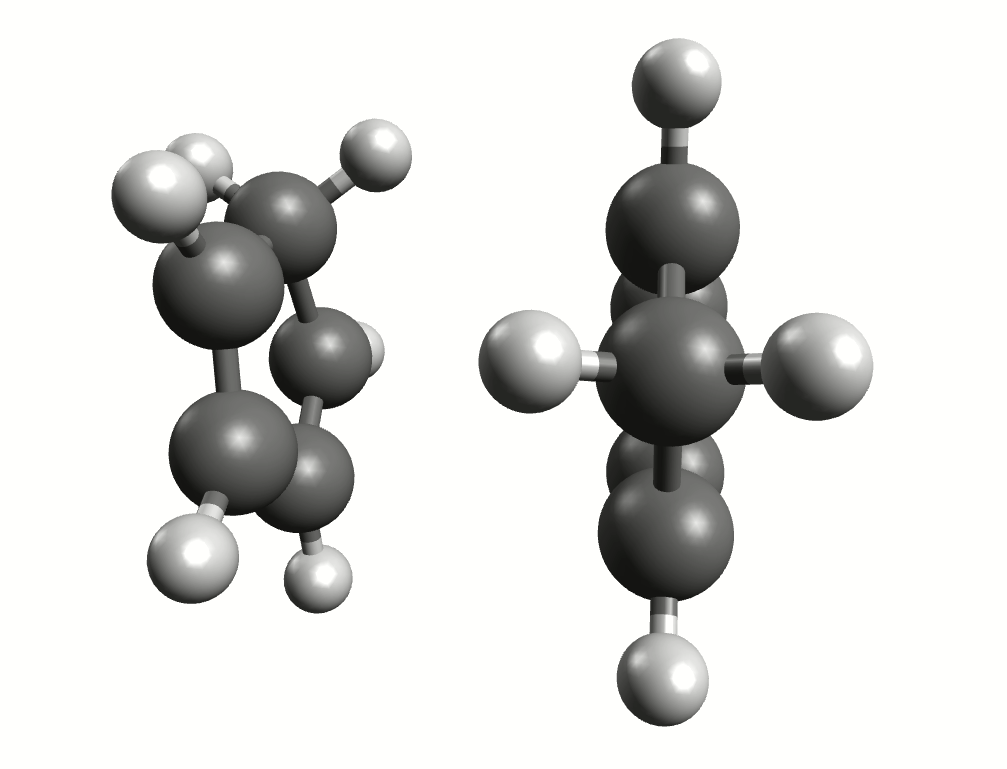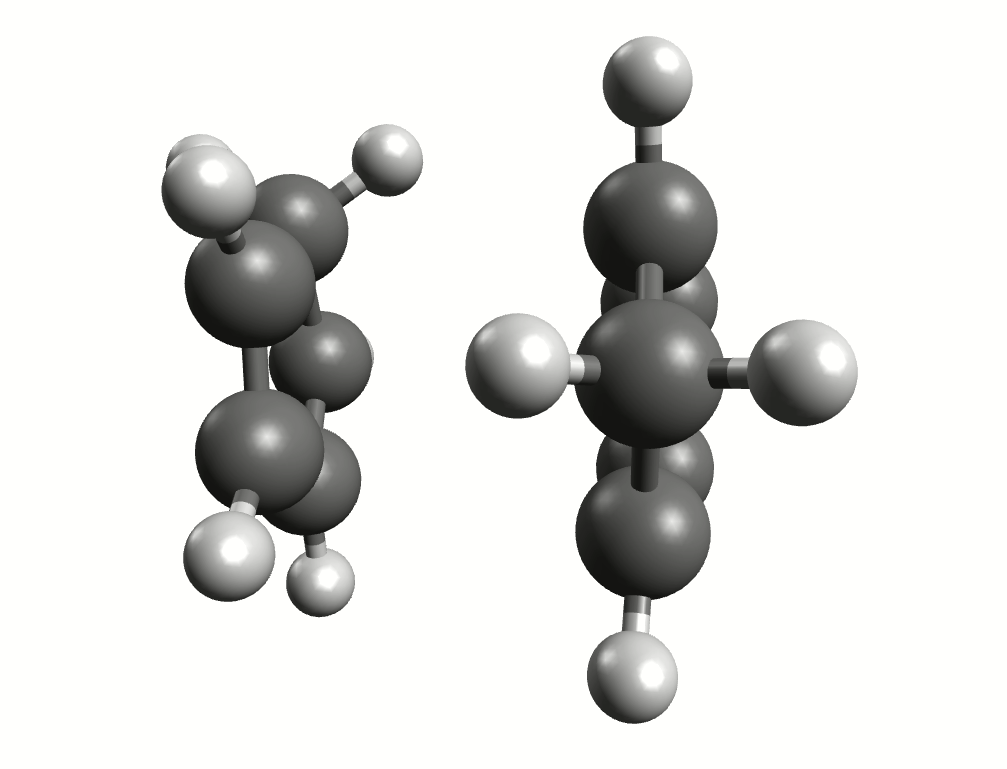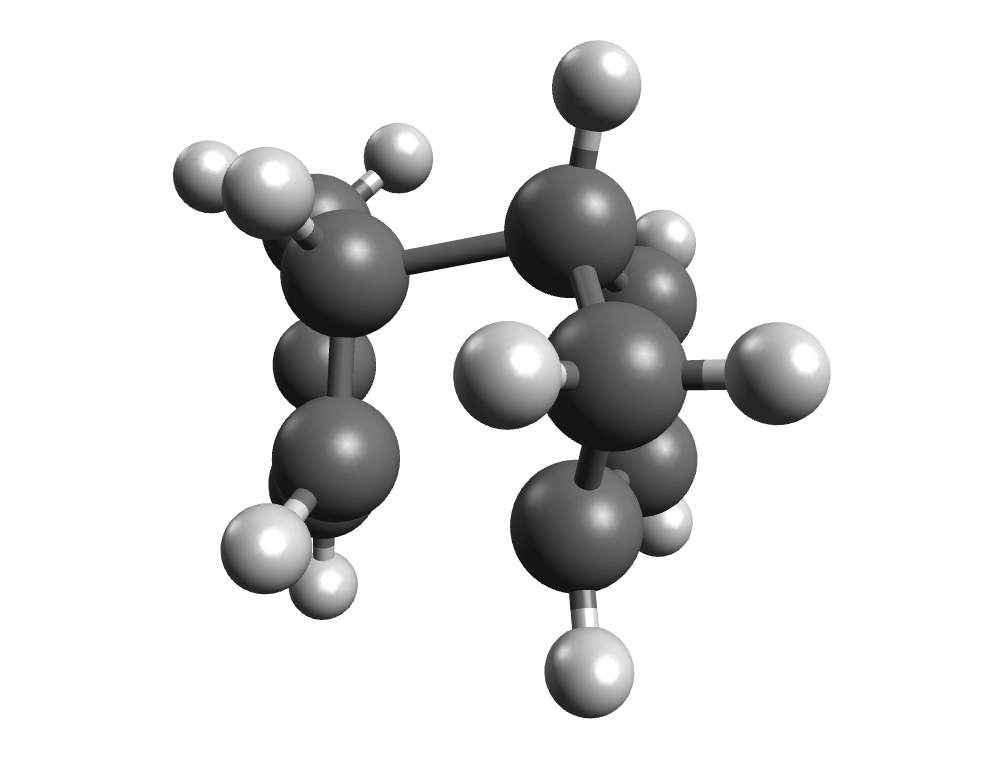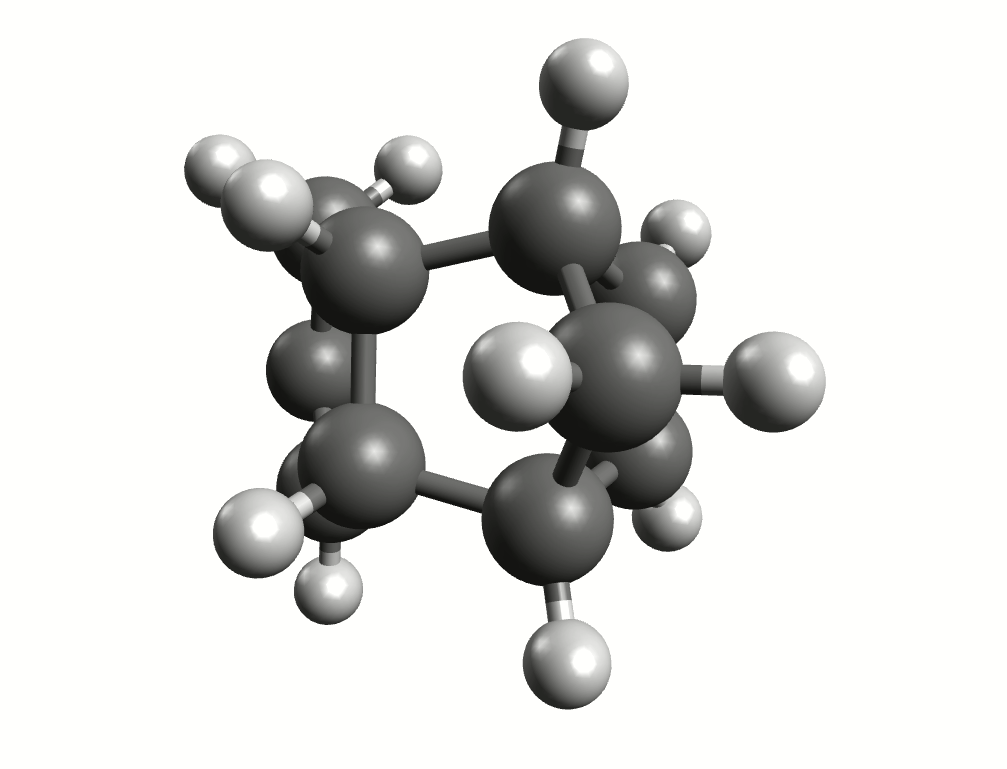
Minimum energy path for the Diels-Alder reaction from the NEB-TS calculation.
The resulting reaction and activation (free) energies are printed again in the summary file as following:
-24.523926 Reaction Energy dE TD wB97X-V def2-TZVP CPCM(Water) 0 1 NEB-TS Summary DFT
21.780152 Activation Energy dE TS wB97X-V def2-TZVP CPCM(Water) 0 1 NEB-TS Summary DFT
-17.070249 Reaction Free Energy dG TD wB97X-V def2-TZVP CPCM(Water) 0 1 NEB-TS Summary DFT
24.855281 Activation Free Energy dG TS wB97X-V def2-TZVP CPCM(Water) 0 1 NEB-TS Summary DFT
Reaction between cyclopentadiene and methylcyclopentadiene - Using a template Hessian
Now that we have the results of the Diels-Alder reaction between the two cyclopentadienes, we can proceed to see how the reactivity changes when we add a methyl group to one of the cyclopentadienes.
Instead of starting the whole process from scratch, we can use the optimized reactant complex, product, and transition state from the previous calculation and modify them accordingly. The modification involves replacing one of the hydrogen atoms attached to one of the carbon atoms with a methyl group. This can be done in any molecular builder tool of your choice. If you are not familiar with it, you can refer to our tutorial on how to use Avogadro. When adding the methyl group, be sure to replace the exact hydrogen in the reactant complex, product, and transition state. This step will keep the numbering consistent in all three systems.
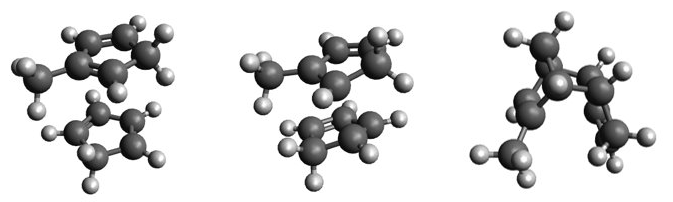
Input structures for Diels-Alder reaction between cyclopentadiene and methylcyclopentadiene: reactants (left), transition state (center), and product (right).
You can save the updated structures in the files DA_reactant2.xyz, DA_product2.xyz and DA_ts2.xyz.
From here, there are two approaches to calculating the activation and reaction (free) energies for this second reaction.
One is to follow the same steps as before, using the reactivity workflow.
In this case, however, you can use the previously prepared transition state structure as a guess for the
transition state using the -tsguess keyword. The WEASEL command would then read:
$ weasel DA_reactant2.xyz -W Reactivity -product DA_product2.xyz -tsguess DA_ts2.xyz
Running this calculation would produce the following results in the summary file:
-24.502095 Reaction Energy dE TD wB97X-V def2-TZVP CPCM(Water) 0 1 NEB-TS Summary DFT
24.679614 Activation Energy dE TS wB97X-V def2-TZVP CPCM(Water) 0 1 NEB-TS Summary DFT
-17.130277 Reaction Free Energy dG TD wB97X-V def2-TZVP CPCM(Water) 0 1 NEB-TS Summary DFT
27.645757 Activation Free Energy dG TS wB97X-V def2-TZVP CPCM(Water) 0 1 NEB-TS Summary DFT
Comparing the results of the Diels-Alder reaction of two cyclopentadienes and a cyclopentadiene with a methylcyclopentadiene, it can be seen that both have a similar reaction (free) energy. However, the activation (free) energy is higher in the case of the cyclopentadiene with methylcyclopentadiene, showing that the methyl group inhibits the reaction.
While this approach is convenient in that WEASEL would provide you directly with the activation and reaction (free) energies, it is not the most computationally efficient way. The reactivity workflow, despite the transition state guess, would perform a full NEB calculation, which is the most time-consuming step, and the transition state guess only makes the search more accurate, not necessarily significantly faster. However, due to the previous calculation of the two cyclopentadienes, you already know your transition state quite accurately and therefore do not really need the NEB calculation. This brings us to the second approach:
To skip the NEB calculation when calculating your activation and reaction (free) energies, you can do three separate calculations. You need to optimize the reactants and product structures using the following WEASEL commands:
$ weasel DA_reactant2.xyz -freq
and:
$ weasel DA_product2.xyz -freq
The -freq is needed to calculate the free energies.
To optimize the transition state you must use the -optts keyword.
To further increase the efficiency of the calculation you can use the Hessian from the previous calculation
(DA_ts.hess) as a template for the current run using the keyword -optts-inputhessian.
This template Hessian strategy can make the process much faster and more accurate.
The complete WEASEL command to compute the transition state would then be:
$ weasel DA_ts2.xyz -optts -optts-inputhessian DA_ts.hess -freq
From the separate results of the reactant, product, and transition state optimizations, you can then manually calculate the reaction and activation (free) energies. The reaction (free) energy must be calculated as the difference between the total (free) energy of the products and the reactants, and the activation (free) energy must be calculated as the difference between the total (free) energy of the transition state and the reactants.
Note
To further simplify the transition state search, you can also use the keywords -optts-activeatoms A [A ...]
or -optts-activeatoms-template A [A ...].
In the first case you have to specify the atom numbers of the active atoms of the transition state,
i.e. the atoms involved in the reaction.
In the second case you have to specify the active atoms of the transition state of the template Hessian
(-optts-inputhessian).
Rotating groups
For reactions, in which larger chemical groups are rotating, or for the comparison of multiple paths (e.g. rotation via two different directions), providing a guess for the TS structure is recommended.
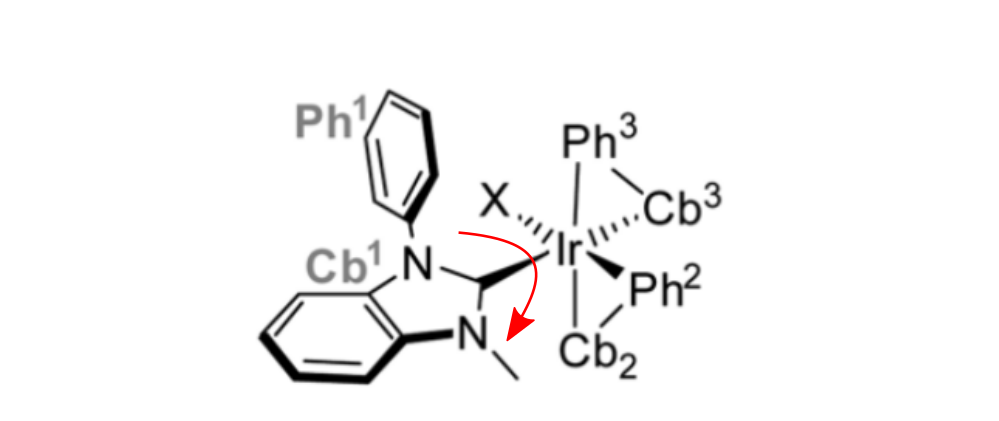
Rotation of a large group around the Ir-C bond.
This TS guess can be a simple "educated guess", and will be used for the generation of the initial path.
It can be provided with the keyword -tsguess"
weasel Ir_HCl_II.xyz -W exploreReactivity -product Ir_HCl_III.xyz -tsguess Ir_TSGuessI.xyz
The exploreReactivity workflow produces the reaction's MEP:
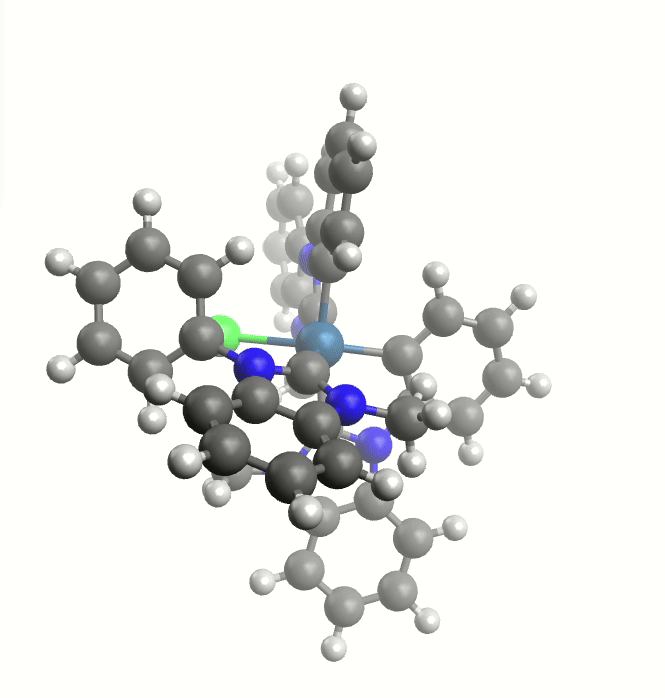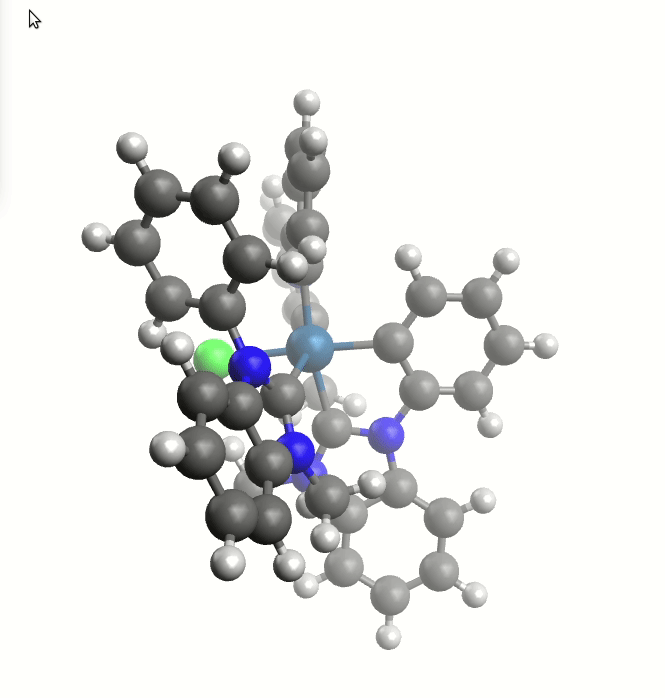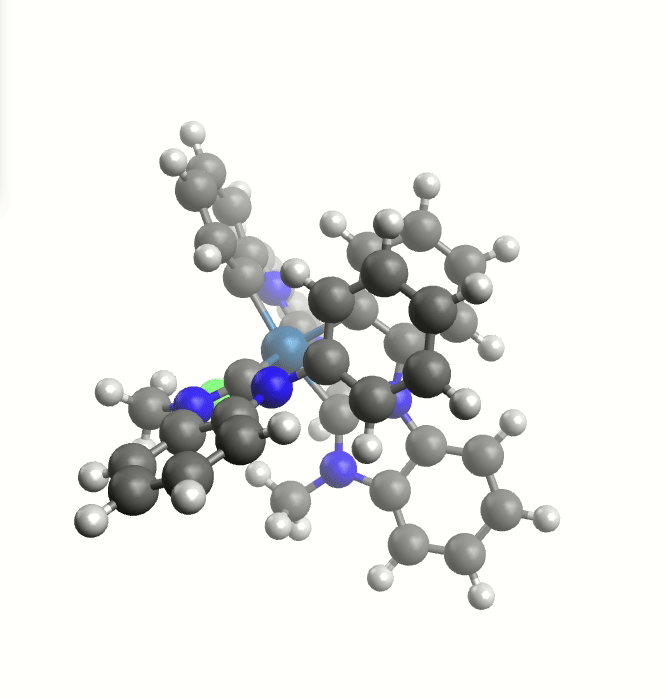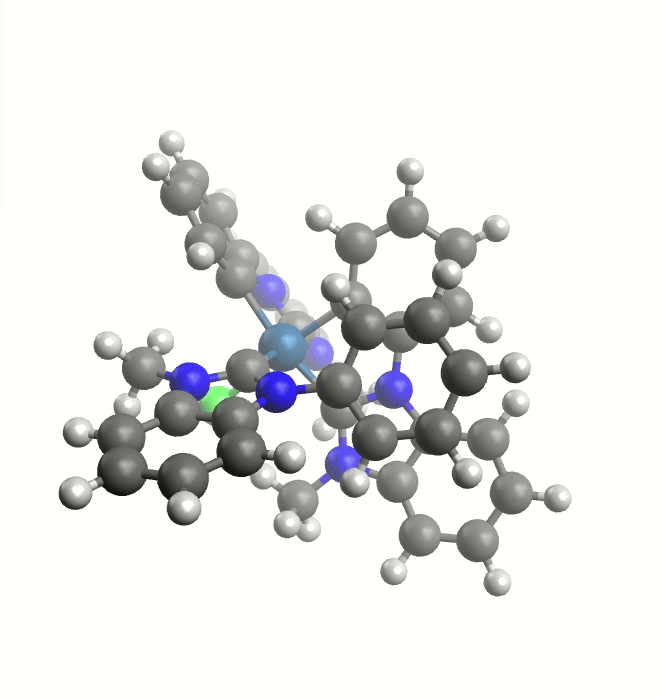
Minimum energy path from the NEB calculation.
TS structure, and activation and reaction free energies:
-10.635936 Reaction Energy dE TD RIJCOSX-B3LYP def2-TZVP(-F) Gas phase 0 1 NEB-TS Summary DFT
16.795442 Activation Energy dE TS RIJCOSX-B3LYP def2-TZVP(-F) Gas phase 0 1 NEB-TS Summary DFT
-9.908759 Reaction Free Energy dG TD RIJCOSX-B3LYP def2-TZVP(-F) Gas phase 0 1 NEB-TS Summary DFT
19.399550 Activation Free Energy dG TS RIJCOSX-B3LYP def2-TZVP(-F) Gas phase 0 1 NEB-TS Summary DFT
Keywords and Remarks
Keywords for reactivity calculations
Keyword |
Description |
|---|---|
|
Calculates the reaction and activation free energy for a reaction between
provided reactant and product.
|
|
Calculates the reaction and activation free energy for a reaction between
provided reactant and product on a lower level of theory.
|
|
File with product structure.
|
|
Find TS with NEB-TS Feature.
|
|
Use guess for minimum energy path in NEB calculation. Guess provided in
ORCA's allxyz file format as input structure file.
|
|
Reactant and product structure should be kept as they are.
|
|
If requested, compute SP and frequency for reactant, TS, and product.
|
|
Use Fast-NEB-TS to boost the convergence. Comes with slightly lower
robustness.
|
|
Use this number of images for the NEB calculation.
|
|
Rerun optimization in case (unwanted) imaginary modes are found with
the frequency analysis.
|
|
Do not rerun optimization in case (unwanted) imaginary modes are found
with the frequency analysis.
|
|
Compute electro- and nucleophilicity.
|
Keywords for transition state calculations
Keyword |
Description |
|---|---|
|
Do TS optimization
|
|
Input hessian of (previously optimized) TS structure. Needs to be an
ORCA Hessian file.
|
|
Active atoms of transition state, i.e. atoms that are involved in reaction.
|
|
Active atoms of transition state of template Hessian (specified via
-optts-inputhessian), i.e. atoms that are involved in reaction. |
|
Do relaxed surface scan and subsequent TS optimization.
|
|
Find TS with NEB-TS Feature.
|
|
If requested, compute SP and frequency only for TS structure.
|
|
File with TS guess structure for finding TS (optional).
|
|
Compute vibrational frequencies, e.g. at the end of a (TS) optimization.
Free energies are automatically calculated.
|
|
Do not compute vibrational frequencies, e.g. at the end of a (TS)
optimization.
|