Host-Guest Docking
In this tutorial we will explore the use of the Docker as a powerful tool for ensembling atoms and molecules into molecular complexes within WEASEL. Assembling individual components into complexes is critical in computational chemistry, allowing for the search and study of different types of complexes, such as transition metal complexes, reaction complexes, and more. Using the Docker in WEASEL, you can easily assemble a complex by providing the different substructures, which can be atoms, ions, or molecules, without needing to know the exact structure of the final system.
In this tutorial, we will show several examples of using the Docker, such as finding a potential complex as a starting point for an organic reaction, which complements another tutorial discussing the reaction pathway or assembling nitrogenoeus base pairs. However, first, the use of the Docker will be explained step by step for the cobalt complex [CpCo(CO)2] as shown below, which was already used as an example in the explicit solvation tutorial.
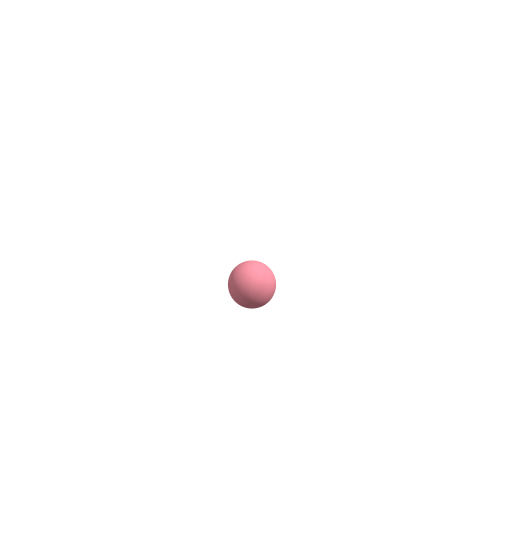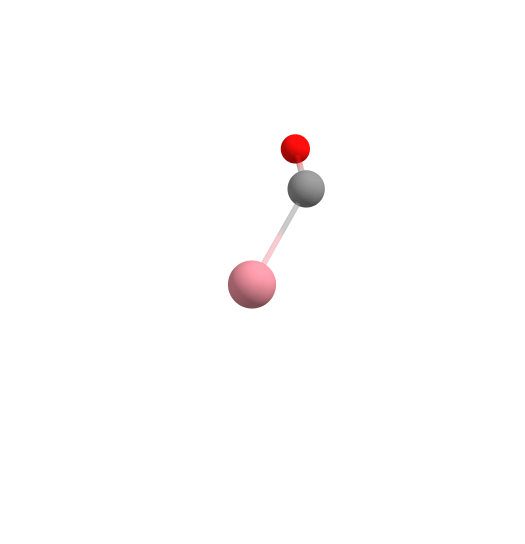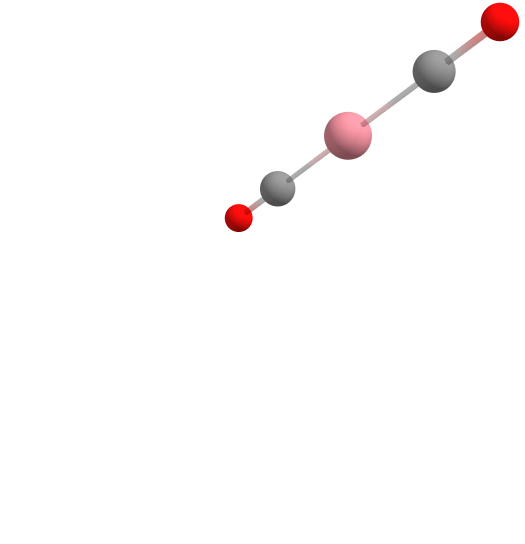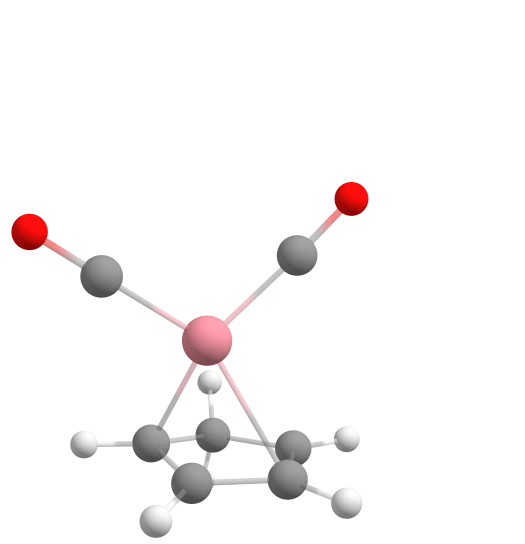
Assembling [CpCo(CO)2] from components using the Docker.
At the end of this tutorial, you will understand how to use the Docker as a useful tool in WEASEL opening up possibilities for incorporating it into further workflows and for studying a wide range of molecular complexes.
How to use the Docker in WEASEL
Let's start the tutorial with the instructions for ensembling the cobalt complex using the provided components of the complex [CpCo(CO) 2] by following the provided steps.
If you want to follow the tutorial, you can download the following XYZ files:
Cp.xyzfor the ligand CpCO.xyzfor carbon monoxideCobalt.xyzfor cobalt
If you create the XYZ files yourself,
make sure that the second line contains the charge and multiplicity information,
as shown in the following example for Cp.xyz:
11
0 1
C 0.25158 -1.00145 -0.63757
[...]
The XYZ coordinates of the different components in relation to each other are chosen arbitrarily.
In the directory where you saved the XYZ files, use the following command to ensemble the complex using the structures
Cp.xyz, CO.xyz, and Cobalt.xyz:
weasel Cobalt.xyz -c 0 -m 4 -dock CO.xyz CO.xyz Cp.xyz
Here, cobalt is the "host", while the molecules that are the desired ligands in the complex are the "guests" docked to the cobalt. In this case, first CO, then CO again, and finally Cp is added to the complex in the docking process. This is shown in the figure above.
Note
The CO molecule needs to be added twice to the complex.
Therefore, its input file, CO.xyz, is specified twice on the command line, while specifying Cp.xyz once.
If all guests need to be added multiple times, you can use the dock-nrepeat keyword followed by the desired number.
However, in this case, specifying -dock-nrepeat 2 to add CO.xyz twice would automatically add Cp.xyz twice as well.
See the example below for a demonstration of how to use the dock-nrepeat keyword.
The keywords -c and -m specify the charge and multiplicity of the cobalt.
They must be specified as given if they are not the default of zero and one, respectively.
However, the charge and multiplicity of the different guests can be specified in the comment line of the input XYZ files, as shown above.
Note
It is also possible to specify the charge and multiplicity of guests by using the keywords -dock-guest-charge and -dock-guest-mult.
Note that these keywords apply the same charge and multiplcity to each guest. If the charge and multiplcity differ between guests,
make sure you specify them in the XYZ files and not directly on the command line.
You can simplify the process by directly specifying the central atom or ion using the -atom option in WEASEL. This approach eliminates the need for an XYZ file containing a single atom. In the presented case you can use the following command:
weasel -atom Co -c 0 -m 4 -dock CO.xyz CO.xyz Cp.xy
In this command, -atom Co directly specifies the central atom as cobalt, without requiring a separate XYZ file.
The -c and -m options again specify the charge and multiplicity of the central atom.
Note
By default, when using the Docker without any additional command line modifications,
the basic WEASEL workflow (Preoptimization, DFT Optimization, SP DFT) is executed after the docking process.
If you just want to use the Docker without the basic workflow,
you can use the keyword -dock-only followed by the guests instead of -dock.
The desired docking process is visualized below.
What the Docker does
The Docker module in sequentially adds the guest molecules to the host in the order specified in the command. In our specific case, it first adds carbon monoxide to cobalt to form the [Co(CO)] complex. Then it adds more carbon monoxide to the previously assembled [Co(CO)] complex, creating the [Co(CO)2] complex. Finally, it incorporates Cp (cyclopentadienyl) to form [CpCo(CO)2].
For each guest molecule addition, the Docker performs a search for multiple potential energy minima on the potential energy surface using XTB, with the new guest added at different positions to the existing structure. It selects the lowest energy minimum and proceeds to add the next guest molecule until the entire complex is fully assembled, as illustrated for each docking step in the figure below.
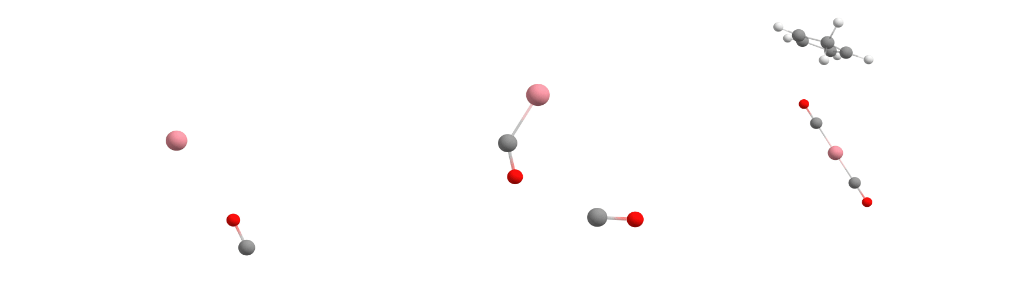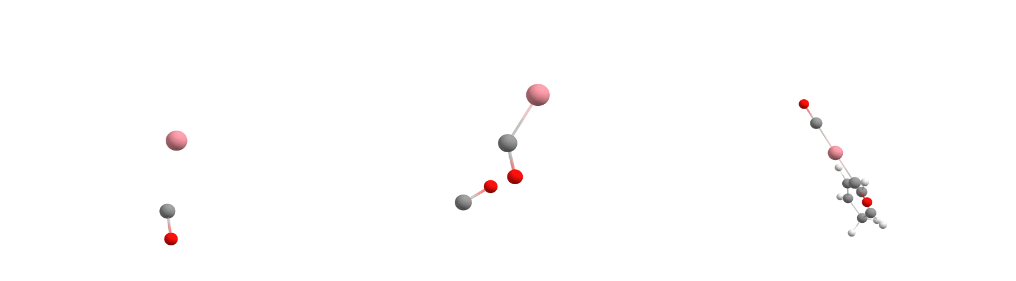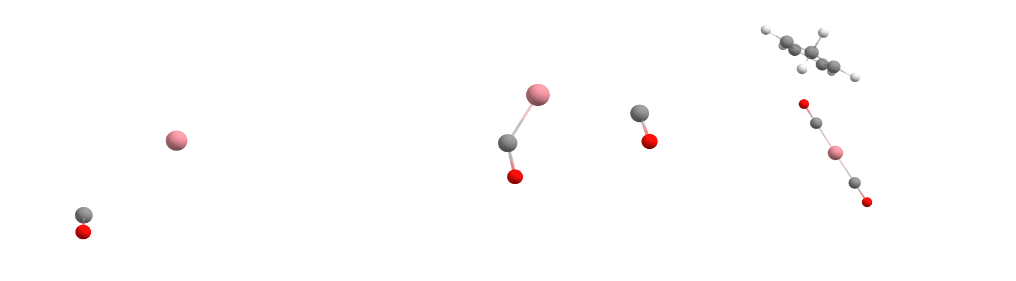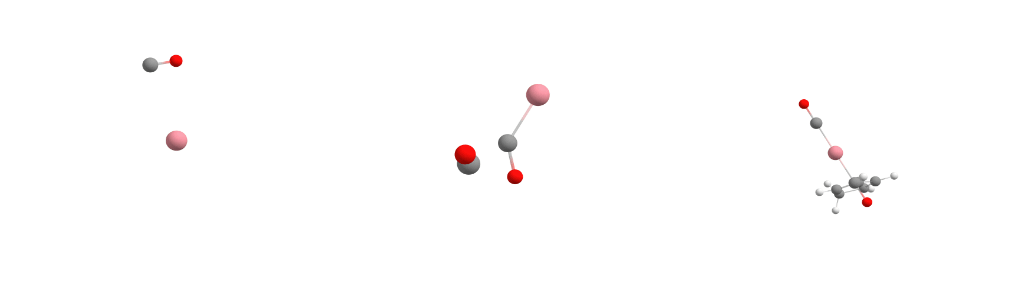
Docking process adding the first CO (left), the second CO (center), and the Cp ligand (right) to the cobalt complex.
The resulting lowest energy structure from the last run is carried forward to all subsequent potential calculation steps.
If the -dock-only option is used, the calculation ends after this step and the final complex structure is obtained.
Note
The Docker in WEASEL may not always find the lowest energy structure because it generates multiple structures at the XTB level and only passes the lowest energy structure to the next docking and finally, to the follow-up calculation steps. Therefore, in cases where multiple conformers are relevant, it may be useful to perform a conformer search after docking. To do so you can run the command:
$ weasel Cobalt.xyz -c 0 -m 4 -dock CO.xyz CO.xyz Cp.xyz -confsearch
However, the standard conformer search in WEASEL may not effectively determine the best binding mode, especially for metal-ligand complexes. In such situations, the optimal binding workflow is recommended as it provides a more appropriate approach to identifying optimal conformer ensembles in metal-ligand complexes. To do so you can run the command:
$ weasel Cobalt.xyz -c 0 -m 4 -dock CO.xyz CO.xyz Cp.xyz -W OptimalBinding
WEASEL output files
For a run in which -dock is requested with no further workflow modification, the output file structure is as follows:
.
├── Cobalt.xyz
└── Cobalt
├── Cobalt_Opt.xyz
├── Cobalt.report
├── Cobalt.results.xyz
├── Cobalt.summary
├── Cobalt_Docking.docker.xyz
├── BuildTopo
│ └── Cobalt_BuildTopo job files
├── Docking
│ └── Cobalt_Docking job files
├── PreOpt
│ └── Cobalt_PreOpt job files
├── Opt
│ └── Cobalt_Opt job files
└── SP_DFT
└── Cobalt_SP_DFT job files
The most important files and a brief description of their contents are listed in the table below.
File |
Description |
|---|---|
Cobalt.results.xyz |
Final optimized host-guest structure after WEASEL basic workflow |
Cobalt_Docking.docker.xyz |
Host-guest structure after docking |
Cobalt.report |
Information about each step of the calculation and information about the
interaction energies and distances between host and guest
|
Cobalt.summary |
Summary of results including XTB level host-guest interaction energies
and total energy from other steps
|
As always, the report file generated by the Docker contains detailed documentation of all calculation steps. Of particular note, however, is the creation of a table in the action section of the Docker. This table serves as a valuable resource, providing a clear overview of the hosts and guests involved in the ensemble process. The table includes information on the charges and multiplicities associated with each component, allowing you to easily verify that they are arranged in the desired order and state as specified. For ease of identification, the guests are assigned numbers based on the order entered on the command line.
== Starting Action 1. ===
== Docking ==
============================
Charges and multiplicities used for host and individual guest(s):
+---------+--------+--------------+
| | Charge | Multiplicity |
+---------+--------+--------------+
| Host | 0 | 4 |
| Guest 1 | 0 | 1 |
| Guest 2 | 0 | 1 |
| Guest 3 | 0 | 1 |
+---------+--------+--------------+
Starting Docking.
Running /share/orca/orca-master-21713-g8eb9fccb9_clang_openblas_haswell_SMD/bin/orca Cobalt_Docking.inp > Cobalt_Docking.out
Docking completed.
In addition, once the docking process is complete and each Guest has successfully bound to the Host, the resulting interaction energy is displayed in a separate table. It is important to note that this interaction energy is calculated at the XTB level, which is the level at which the docking process takes place. Interaction energies obtained at this level may not be as accurate as those calculated at higher levels of theory. If necessary, it is recommended to recalculate the interaction energies at a higher level of theory to improve the accuracy.
Building topology of host-guest assemblies.
Host-guest complex with 3 sequentially added guest(s) read from: Docking/Cobalt_Docking.docker.xyz
Interaction energies with different numbers of guests:
+-------------+------------------------+--------+--------------+
| # of guests | Int. Energy [kcal/mol] | Charge | Multiplicity |
+-------------+------------------------+--------+--------------+
| 1 | -64.224021 | 0 | 4 |
| 2 | -76.057243 | 0 | 4 |
| 3 | -41.474756 | 0 | 4 |
+-------------+------------------------+--------+--------------+
Writing a xyz file with the final host-guest complex to the mainjob dir: Cobalt_Docking.docker.xyz
It is important to emphasize that the interaction energy given in the table represents the energy between each newly added guest and the host-guest complex. This complex includes all previously docked guests. To illustrate, in the scenario provided, the interaction energy of guest #3 would reflect the interaction energy between the Cp molecule and the cobalt complex, which already includes two previously docked CO molecules.
After completing the additional calculation steps, namely in the standard -dock process after the DFT optimization and SP calculation,
as well as in the -dock-only process immediately after the docking process,
another table is generated and included in the report file.
This table provides information about the distances between the host and the atoms within the guests that are closest to the host in the
final geometry. In the standard -dock case, this table is generated after the DFT optimization,
because the DFT bond lengths tend to be more reasonable compared to the XTB bond lengths as defined within the Docker itself.
========= Starting Action 5. =========
== Calculating Host-Guest Distances ==
========================================
Distances of bonded Host-Guest atoms:
+-------------+---------------+---------+--------------+----------------+---------------------+
| AtomID Host | Atomtype Host | GuestID | AtomID Guest | Atomtype Guest | Distance [Angstrom] |
+-------------+---------------+---------+--------------+----------------+---------------------+
| 1 | Co | 1 | 2 | C | 1.8280 |
| 1 | Co | 2 | 4 | C | 1.8227 |
| 1 | Co | 3 | 6 | C | 2.7580 |
| 1 | Co | 3 | 7 | C | 2.8028 |
| 1 | Co | 3 | 9 | C | 2.1719 |
| 1 | Co | 3 | 11 | C | 2.0829 |
| 1 | Co | 3 | 13 | C | 2.4063 |
+-------------+---------------+---------+--------------+----------------+---------------------+
Within the table you will find the following details: the guest number to which the atom belongs, the atom ID within the final complex, the atom type, and the corresponding distance measured in angstroms. This comprehensive table allows for easy reference and analysis, allowing you to evaluate the proximity and interactions betweenthe host and the atoms in the closest vicinity to the guests.
As usual, the summary file contains the total energies on the different levels of theory calculated for the generated complex, as shown below. In addition, the interaction energies are also included, as already given in the report file.
Energy [kcal/mol] / Value Type Calculation type Method Basis set Solvent Charge Multiplicity Further tags
-76.057243 Int. Energy Docker XTB None ALPB(Water) 0 4 Host-Guest #2
-64.224021 Int. Energy Docker XTB None ALPB(Water) 0 4 Host-Guest #1
-41.474756 Int. Energy Docker XTB None ALPB(Water) 0 4 Host-Guest #3
-18607.870694 SP-Energy PreOpt XTB None ALPB(H2O) 0 4
-1131710.856648 SP-Energy Opt r2SCAN-3c None CPCM(Water) 0 4
-354790.751225 SP-Energy SP_DFT wB97X-V def2-TZVP CPCM(Water) 0 4
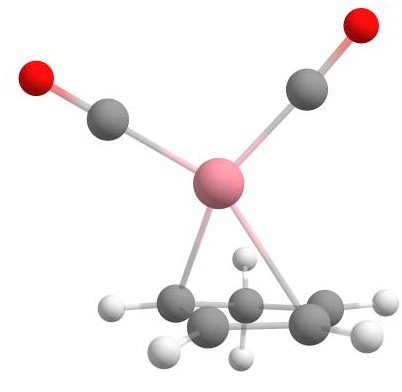
Final structure of the cobalt complex [CpCo(CO)2].
Different examples using the Docker
A metal-ligand complex with repeating ligands
Another example of a metal-ligand complex that can be assembled using the Docker is [Ni(en)2Cl2]0.
Since the two ligands are both twice in the complex, you can use the keyword -dock-nrepeat in combination with the Docker.
To run the calculation,
you can download the XYZ coordinate files en.xyz for ethylenediamine (en) and
cl.xyz for the chloride ion.
In case you create the files yourself make sure that you specified the charge of -1 and the multiplicity (1)
for the chloride ion in the XYZ file.
Then the following WEASEL command can be executed with the charge and multiplicity of the nickel ion specified by -c 2 -m 3:
weasel -atom Ni -c 2 -m 3 -dock-only cl.xyz en.xyz -dock-nrepeat 2
The resulting assembly of the structure with the Docker is shown below.

Assembling [Ni(en)2Cl2]0 from components using the Docker.
A metal ion in water
Let's explore an example where a metal ion, specifically manganese (Mn2+), is to be octahedrally coordinated with water molecules. The desired docking process for the example is visualized below.

Assembling [Mn(H2O)6]2+ from components using the Docker.
To create this complex we can use the following command:
weasel -atom Mn -c 2 -m 6 -dock h2o.xyz -dock-nrepeat 6
If you have read the tutorial on explicit solvation,
you may recall that WEASEL includes the docking utility for one of the two explicit solvation models.
In this sense, this approach is similar to explicit solvation.
However, the main difference lies in the level at which the Docker is used.
The Docker provides improved accuracy compared to the solvation model in the docking process.
However, the level of sophistication of the docking can be changed for the explicit solvation
and the Docker itself with the keyword -dock-level {normal, quick, screening, complete}.
Since there are various scenarios where it can be advantageous to introduce water molecules into a complex as a guest
molecule at a sophisticated level, WEASEL provides a special workflow "Docking-Water" that allows the addition of one or
more water molecules using the standard docking level. The workflow works exactly like -dock,
but without the need to provide the input file structure of the water molecule as a guest.
Using the docking-water workflow, the desired example of Mn2+ with six water molecules can be achieved in
the same way using the following command:
weasel -atom Mn -c 2 -m 6 -W Docking-Water -dock-nrepeat 6
By using this command, the Docker efficiently incorporates the water molecules into the system.
Assembling initial structures for reactions
So far, we have explored the ensembling of metal-ligand complexes using the Docker. However, the Docker has a wide range of applications beyond these specific cases. One such application is the search for reasonable reactant complexes to investigate reaction pathways. For example, let's consider an example involving reactants for a potential Diels-Alder reaction. To find the correct reaction mechanism, we must first determine how the reactants must be positioned with respect to each other.
We are going to examine the Diels-Alder reaction between two cyclopentadiene molecules, which XYZ coordinates (cyclopentadiene.xyz)
are given below:
11
0 1
H -6.49649 0.37554 0.87955
C -6.78843 -0.02322 -0.09623
H -7.60757 0.57116 -0.51107
C -5.61976 -0.06926 -1.03644
C -5.38285 -1.33842 -1.40424
C -6.33844 -2.20605 -0.76280
C -7.16292 -1.47333 -0.00100
H -6.36726 -3.27748 -0.89048
H -7.97659 -1.85760 0.59503
H -5.06049 0.79464 -1.36177
H -4.60603 -1.67738 -2.07257
By designating one reactant as the host and the other as the guest, we can use the Docker to ensemble a complex of two cyclopentadiene molecules:
weasel cyclopentadiene.xyz -dock-only cyclopentadiene.xyz
The result is shown in the figure below.
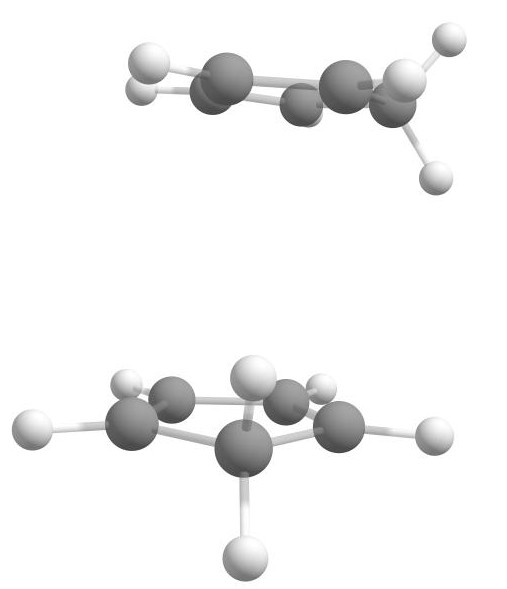
Reactant complex of two cyclopentadiene molecules for Diels-Alder reaction.
The resulting complex shows that Docker has already positioned the relevant components appropriately, with the double bonds of the two reactants positioned in close proximity to each other. This arrangement suggests that the Diels-Alder reaction can potentially occur.
If you want to learn how to calculate reaction pathways with WEASEL, you can read the reactivity workflow tutorial. Structures for use in other workflows can be assembled in the same way.
Assembling of nitrogenous base pairs
Another example of the use of the Docker is the creation of nitrogenous base pairs, which are essential for the DNA double helix.
Specifically, we will use the example of adenine (A) and thymine (T). You can use the XYZ coordinate below to perform the calculation.
For thymine, you can create the XYZ file thymine.xyz with the coordinates:
15
0 1
O -1.51930 1.80670 -0.00090
O 2.83940 0.29130 -0.00070
N 0.66430 1.06230 0.00110
N 1.11480 -1.23160 -0.00020
C -1.16120 -0.54320 0.00020
C -0.71290 0.88020 0.00040
C -0.22600 -1.50130 -0.00010
C -2.63000 -0.81890 -0.00010
C 1.63100 0.05450 0.00030
H -0.47870 -2.55530 -0.00040
H 1.00140 2.02090 0.00110
H 1.75640 -2.01910 -0.00090
H -3.10040 -0.38600 -0.88890
H -3.10060 -0.38620 0.88870
H -2.84330 -1.89310 -0.00030
And for adenine, you can use the following coordinates to create the XYZ file adenine.xyz:
15
0 1
N -1.19900 -1.39970 0.00000
N -2.07520 0.64990 0.00000
N 0.03370 1.85940 -0.00010
N 1.99980 0.41500 0.00000
N 1.82410 -1.97600 0.00000
C -0.13590 -0.54210 0.00010
C -0.70560 0.72410 0.00010
C 1.23590 -0.70040 0.00000
C -2.33860 -0.63940 0.00000
C 1.36070 1.60920 0.00000
H -1.15660 -2.41030 -0.00020
H -3.32250 -1.08640 -0.00010
H 2.00340 2.48310 -0.00010
H 2.83220 -2.05280 0.00030
H 1.25030 -2.80820 0.00040
You can run the WEASEL command as shown below to use the Docker:
weasel thymine.xyz -dock-only adenine.xyz
Alternatively, you can use -dock if you want to run the basic WEASEL workflow after docking as described above.
The resulting nitrogenous base pair is shown in the figure below.
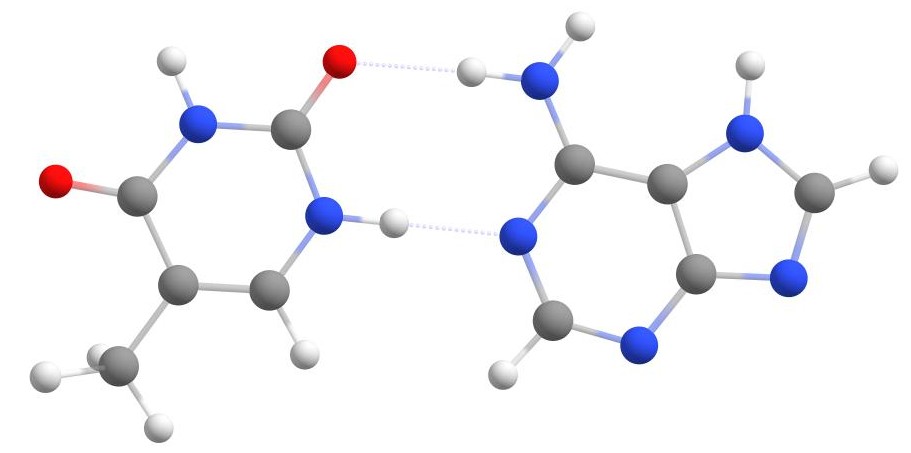
Thymine and adenine base pair.
Note
Studying the interaction energy of the two bases might be interesting, for example. To do this, you can run the interaction workflow after the Docker or the host-guest interaction workflow.
A β-Cyclodextrin complex
As a final example, we will demonstrate the creation of a complex between β-cyclodextrin and p-cresol. Due to the complexity of β-cyclodextrin, we will use its SMILES string as a more compact input. The SMILES string looks like this:
OC[C@H]1O[C@@H]2O[C@H]3[C@H](O)[C@@H](O)[C@@H](O[C@H]4[C@H](O)[C@@H](O)[C@@H](O[C@H]5[C@H](O)[C@@H](O)[C@@H](O[C@H]6[C@H](O)[C@@H](O)[C@@H](O[C@H]7[C@H](O)[C@@H](O)[C@@H](O[C@H]8[C@H](O)[C@@H](O)[C@@H](O[C@H]1[C@H(O)[C@H]2O)O[C@@H]8CO)O[C@@H]7CO)O[C@@H]6CO)O[C@@H]5CO)O[C@@H]4CO)O[C@@H]3CO
You can use this SMILES string directly on the command line, or save it in a file named, for example, bcd.smi.
However, the guest structure, p-cresol, should be included as an XYZ input file,
since we need the comment line of the file to specify its charge and multiplicity.
The XYZ file, called pcresol.xyz below, should contain the following XYZ coordinates:
22
0 1
C -0.566000000 1.310000000 0.343000000
C 0.609000000 -0.810000000 1.066000000
C -0.588000000 0.802000000 -1.096000000
C 0.583000000 -1.312000000 -0.375000000
C -0.553000000 0.152000000 1.350000000
C 0.566000000 -0.157000000 -1.374000000
C -0.476000000 0.682000000 2.781000000
O 0.424000000 -0.668000000 -2.695000000
H 0.320000000 1.942000000 0.489000000
H -1.442000000 1.946000000 0.516000000
H 0.562000000 -1.667000000 1.748000000
H 1.565000000 -0.306000000 1.255000000
H -1.544000000 0.301000000 -1.299000000
H -0.544000000 1.648000000 -1.793000000
H 1.454000000 -1.952000000 -0.563000000
H -0.294000000 -1.952000000 -0.533000000
H -1.494000000 -0.405000000 1.248000000
H 1.515000000 0.391000000 -1.328000000
H 0.449000000 1.241000000 2.918000000
H -0.498000000 -0.149000000 3.484000000
H -1.322000000 1.339000000 2.977000000
H 1.170000000 -1.271000000 -2.841000000
To perform the calculation using the Docker feature, run the following command
weasel bcd.smi -dock-only pcresol.xyz
Remember, if you want to continue with the basic workflow after docking,
you can replace -dock-only with the -dock keyword in the command.
The docker will handle the interaction between β-cyclodextrin and p-cresol, and the resulting structure will be generated. The figure below shows the final structure after docking.
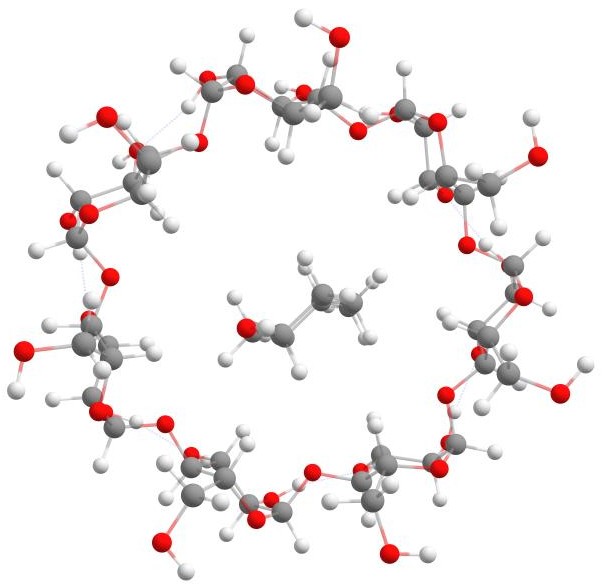
β-Cyclodextrin with p-cresol.
Remarks and keywords
Keyword |
Description |
|---|---|
|
Determine best binding position of a single or
multiple guests to a host system. GUEST is xyzfile
with one or more guests. Multiple xyzfile can be
specified, by separating them with spaces. The charge
and multiplicity are of each invidual guest is read
from the comment line of the entry in the xyzfile(s).
Therefore, the comment line must contain exactly two
integers, where the first is the charge and second one
the multiplicity.
|
|
Disable docking procedure.
|
|
This keyword has the same function as '-dock', but it
also turns off any other workflow.
|
|
Add guest(s) N times in docking process. Guests are
read repeated in the order the order they were read.
|
|
Set total charge for every guest structure. By default
charge is read from first column of XYZ comment line (if
present).
|
|
Set multiplicity for every guest structure. By default
multiplicity is read from second column of XYZ comment
line (if present).
|
-dock-level{normal, quick, screening, complete} |
Level of sophistication used for docking. |
|
Bonding factor N (e.g., 1.5), by which sum of radii of
host and guest is scaled. If intermolecular distance is
below this value, host and guest are considered bound.
|
|
Keep geometry of host fixed during docking. |
|
Do not keep geometry of host fixed during docking. |