A conformer represents a unique arrangement of the atoms of a molecule,
characterized by different spatial orientations and configurations of the atoms while maintaining a particular connectivity between them.
Conformers can differ in their energy levels and stability, and they can interconvert through molecular motions such as rotation,
vibration, or even more complex changes.
Exploring the different conformers of a molecule and searching for the lowest energy conformer or set of conformers can be useful for
several reasons. The WEASEL conformer search workflow provides a convenient way to find them. This workflow is useful in itself,
but it is also the basis for several other workflows that explore the properties and reactivity of a molecule in different conformers.
For example, it is possible to calculate different spectra such as NMR, IR, or
UV/Vis and CD spectra for a conformer ensemble.
The WEASEL conformer search workflow is also the basis for several other chemical search workflows,
such as the anion and protomer searcher or the tautomer searcher,
to name a few.
For now, let us focus on the basics of the conformer search workflow by taking a look at the conformer ensemble of limonene.
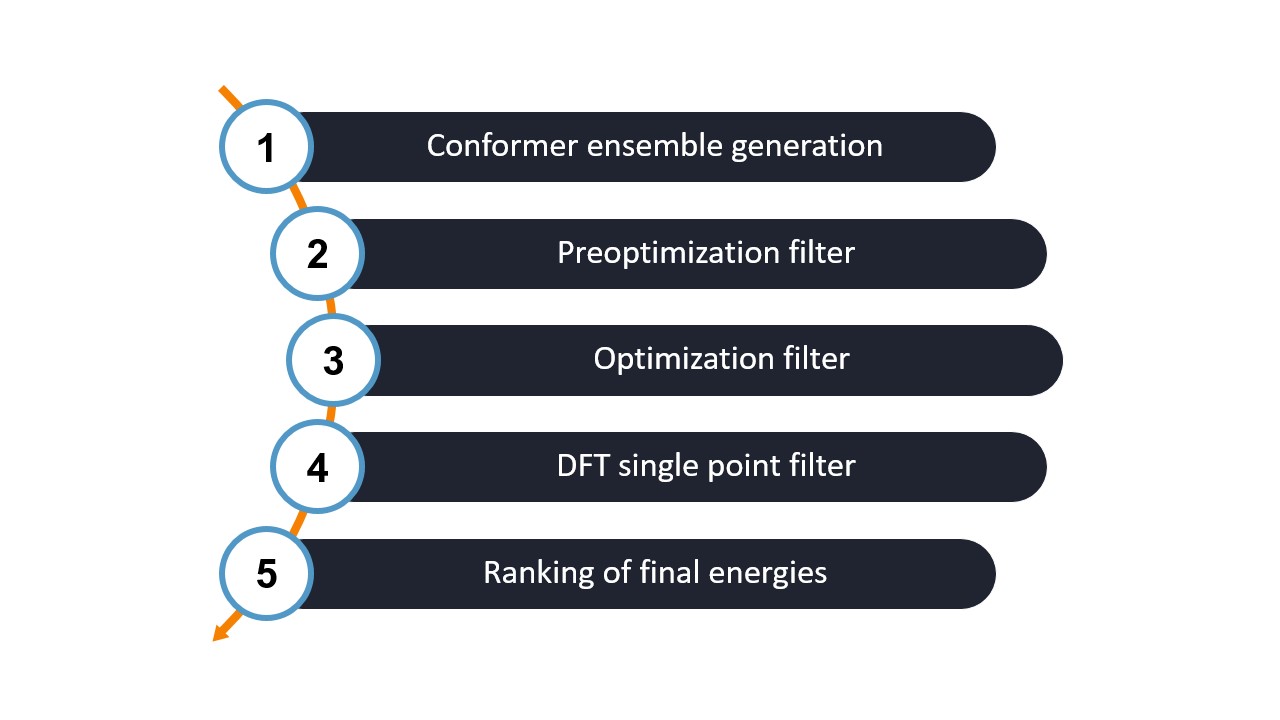
The WEASEL conformational search workflow consists of several filtering steps to obtain a conformer ensemble for a system.
Let's walk through each step of the workflow:
Step 1: Conformer generation
This step generates a set of initial conformers for the system. The conformers are generated using CREST.
The energy filter window for this step is 6.0 kcal/mol.
This means that WEASEL ranks the resulting conformers by energy, and only conformers that differ by 6.0 kcal/mol or
less from the lowest energy conformer are retained in the ensemble. This ensemble is passed to the next step.
Note
You can change the method used to generate the conformer ensemble by applying the keyword -conf-gen-method followed by the desired
method. The available methods are READ, CREST, CRESTFF, RDKIT, CREST-RDKIT, CRESTFF-RDKIT, GOAT, GOATFF.
The next three filtering steps (step 2 to step 4) consist of
a preoptimization step, followed by an optimization and a final single point DFT calculation using the same methods as in the
basic workflow.
Note
You can modify the default methods to suit your needs and preferences using the here provided keywords.
Note that they are different to those used to customize the basic Workflow.
Step 2: Preoptimization and clustering
Preoptimization with XTB using ORCA is performed on the ensemble with an energy filter window of 6.0 kcal/mol.
This step refines the generated conformations and prepares them for further optimization.
In addition, a fine clustering step is applied to group similar conformers together.
Step 3: Optimization
In this step, optimization is performed using ORCA with the r2SCAN-3c method.
The energy filter window is set to 5.0 kcal/mol. The optimization ensures that the conformations reach stable energy minima.
Step 4: Single point energy Calculation
After optimization, the single point energy is calculated using the wB97X-V method with the def2-TZVP basis set with ORCA.
The energy filter window is set to 4.0 kcal/mol. This calculation provides more accurate energy values for the conformers.
Note
If you want even more accurate energies, you can follow up with a single point calculation using a wavefunction based method.
To invoke this additional calculation step, you can use the keyword -conf-spwf.
As mentioned above, throughout the entire workflow, all high-energy structures outside of a specified energy window are eliminated
in order to retain only relevant conformers in each step.
In addition, duplicates are removed to ensure that only unique conformers are retained.
The table provided summarizes the default conditions associated with each filtering step.
Filters applied after each step of the conformation search together with their default values.
Step
Relative energy
(kcal/mol)
Remove
Identical
Conformer
Remove
Identical
Rotamer
Ensemble generation
no
yes
yes
Preoptimization Filter
6
yes
yes
Optimization Filter
5
yes
yes
DFT SP energy Filter
4
no
no
Wavefunction SP energy Filter
3
no
no
Note
If you want to change the energy windows from step 1 to step 4,
you can do so by using the following keywords followed by the desired energy in [kcal/mol]:
conformer generation - -conf-gen-enrange, preoptimization - -conf-preopt-enrange,
optimization: -conf-preopt-enrange, SP calculation - -conf-spdft-enrange.
See also here for more information.
Step 5: Ranking of final energies
After generating the final ensemble, WEASEL evaluates the energies of its conformers and produces a summary file showing the ranking
of these energies. In the following section, the summary file and the other output files will be explained in more detail.
Note
By default the conformer search workflow is performed in the solvent water.
You can switch to a different solvent by using the keyword -solvent [solvent].
The list of solvents can be found here.
Before discussing the individual steps of the conformational search further below, let us first have a look at
the results of the confsearch workflow. The calculation produces the following files:
The most important files and a brief description of their contents are listed in the table below.
File
Description
limonene_lowestConf.xyz
lowest-energy conformer
limonene.ensemble.xyz
conformer ensemble
limonene_confsearch.report
Output file of the WEASEL run
limonene_confsearch.summary
Summary file for the WEASEL run
The final conformer ensemble is stored in a multi-xyz file ('limonene_ConfSearch.results.xyz')
along with the single point DFT calculated energy of each individual conformer.
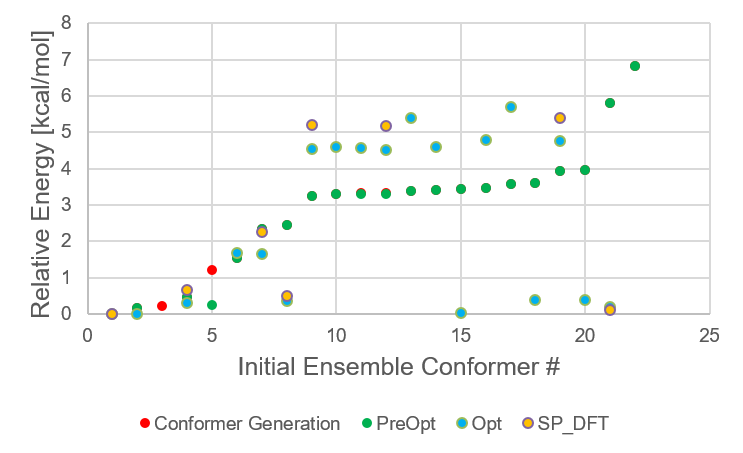
During the filtering steps a lot of information was collected, which was stored in the summary file. There, for each
filtering step, we find the energies of all conformers that have survived up to that step:
The last column of the summary file contains the ID of the conformers from the initial conformer ensemble.
Note
With each filtering step, more and more conformers are filtered out, thus fewer and fewer conformer IDs are
available from step to step. The numbering of these conformers in the last column is not in ascending order, but in the order of
their relative energies in the very first step.
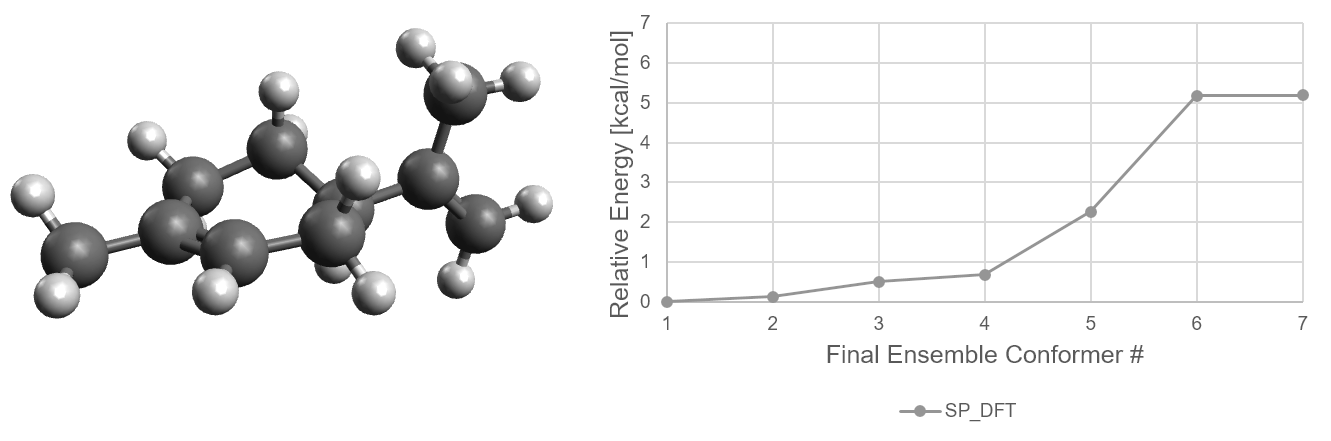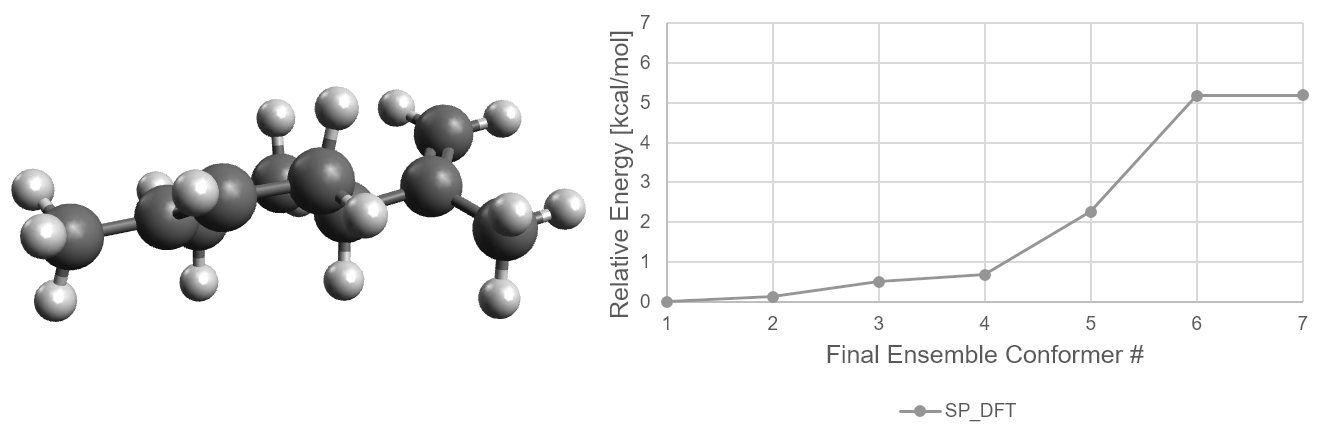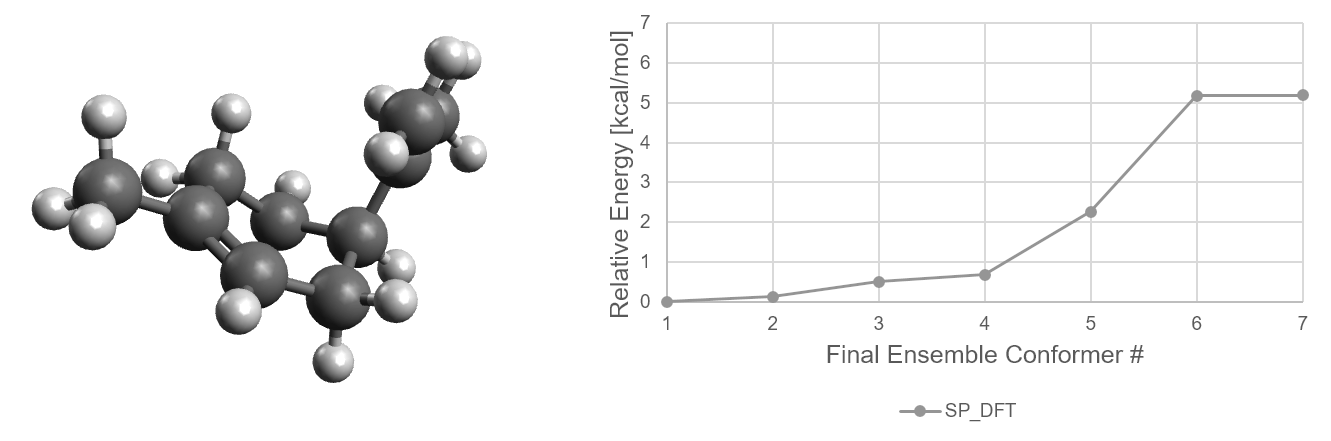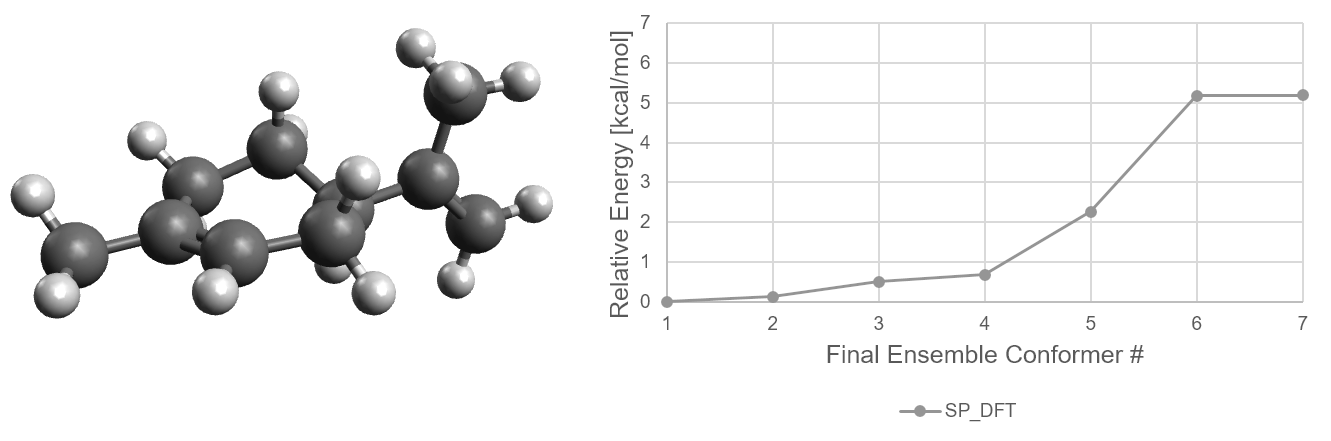
Now let us make use of that data. The following graph shows how the relative stability of each conformer evolves with
increasingly accurate methods.
Energetic distribution of limonene conformer ensemble in initial ensemble numbering scheme.
For limonene, the lowest energy conformer remains the same through all filter steps. The higher energy
conformers change their relative energies more significantly.
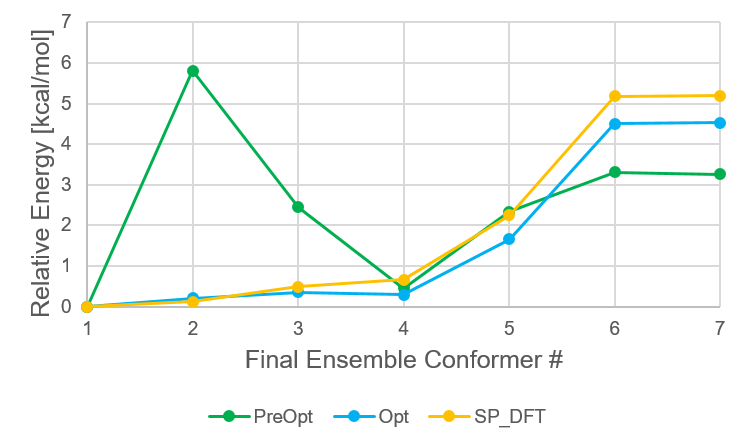
The following figure shows the ensemble of conformers that survived to the SP_DFT step, and the relative
energies of each of these conformers at each of the filtering steps. From the preoptimization step to the
optimization step, the relative energies and even the order can change quite drastically. However, from the
Opt step to the SP_DFT step, the results for the limonene conformers are quite similar.
Energetic distribution of limonene conformer ensemble in final ensemble numbering scheme.
Method for providing the initial conformer ensemble, which is then
refined using the subsequent filtering steps. Available options are CREST,
CRESTFF, GOAT, GOATFF, RDKIT and READ. The READ option allows the user
to provide an initial conformer ensemble, e.g. from a different conformer
generator. If the READ option is requested, the initial conformer
ensemble has to be provided via the structure file as a multi-xyz file.
-conf-gen-maxnconfINT
Maximum number of conformers selected for the next steps. The first
INT structures from the initially generated or provided conformer
ensemble are considered. The remaining ones are discarded.
-conf-gen-enrangeREAL
Energy filter in [kcal/mol]. Energies are computed on GFN2-xTB level.
Only conformers with a relative energy of less than REAL compared
to the current lowest-energy conformer are considered. The remaining
ones are discarded. This is not used for the READ conformer generation
method.
-conf-torsionfilterINT1INT2INT3INT4
Use an additional dihedral filter on generated or provided initial conformer
ensemble. Default is to not use it. If the four atoms for the definition of
the torsion angle are provided via -conf-torsionfilter, the filtering step
is switched on.
-conf-torsionfilter-rangeREAL1REAL2
Only those conformers, for which this torsion is in the range between
REAL1 and REAL2, are considered for the next steps. Torsion angles
in degrees.
Keywords for preoptimization filter
Keyword
Description
-conf-preopt
Use preoptimization step. Default is true.
-conf-preopt-enrangeREAL
Energy filter in [kcal/mol]. Only conformers with a relative energy of
less than REAL compared to the current lowest-energy conformer are
considered. The remaining ones are discarded.
Keywords for optimization filter
Keyword
Description
-conf-opt
Use optimization step. Default is true.
-conf-opt-enrangeREAL
Energy filter in [kcal/mol]. Only conformers with a relative energy of
less than REAL compared to the current lowest-energy conformer are
considered. The remaining ones are discarded.
-conf-gibbscorrection
Run frequency calculation after optimization and use the Gibbs correction
for the optimization, DFT and wavefunction Single Point filter steps.
Default is false.
Keywords for DFT single point filter
Keyword
Description
-conf-spdft
Use DFT SP energy step. Default is true.
-conf-spdft-enrangeREAL
Energy filter in [kcal/mol]. Only conformers with a relative energy
of less than REAL compared to the current lowest-energy conformer
are considered. The remaining ones are discarded.
Keywords for wavefunction single point filter
Keyword
Description
-conf-spwf
Use wavefunction SP energy step. Default is false
-conf-spwf-enrangeREAL
Energy filter in [kcal/mol]. Only conformers with a relative energy of
less than REAL compared to the current lowest-energy conformer are
considered. The remaining ones are discarded.
The default method for the wavefunction single point filter is DLPNO-CCSD(T) with def2-TZVP basis set,
and needs to be modified via the workflow file in the CONFORMATIONAL_SEARCH section:
[CONFORMATIONAL_SEARCH]
# Options: see weasel -h
SP_WF_Method = DLPNO-CCSD(T)
# Options: see [SP_WF] Basis
SP_WF_Basis = def2-TZVP
Keywords for changing the maximum number of conformers
Keyword
Description
-conf-maxnconfINT
The maximum number of conformers that is stored in the ensemble file.
Keywords for clustering conformers
Apply an agglomerative hierarchical clustering with complete linkage after the optimization step. The distance matrix is composed of the RMSDs to the lowest energy structure augmented by energy information.
If done, a folder named CLUSTERS will be created inside the Opt folder where each individual cluster can be visualized.
Keyword
Description
-conf-cluster
Apply the clustering after the optimization step.
-conf-cluster-modeOPTION
Mode of clustering. Choose between fine (default) and coarse for
more compression of the data.
-conf-cluster-nmaxINT
Define an arbitrary maximum number of clusters. The default is -1,
meaning that it will be defined automatically.
-conf-cluster-elevelOPTION
Select when to apply the clustering, after the PreOpt step (default) or