Ion Mobility Collision Cross Sections
The ion mobility collision cross section (CCS) is a value that refers to the area of a particle (atom or molecule) where interactions with another particle can take place. CCS values are routinely measured using ion mobility spectrometry (IMS), and are of significant interest in areas like metabolomics, in which it is used to obtain information about the lowest energy conformers.
How to run the calculation
Inside WEASEL, all necessary steps to compute CCS values are done by one of three CCS workflows.
Chosing the right workflow depends on the chemical properties of the input structure.
In the following, we will use Ethiprole as an example, for which we took the structure from
PubChem (as a SDF file).
If you want to run the structure by yourself, you can also use its SMILES string with the
keyword -smiles CCS(=O)C1=C(N(N=C1C#N)C2=C(C=C(C=C2Cl)C(F)(F)F)Cl)N.
It is usually the case that the CCS is computed for charged forms of neutral molecules, either by protonating/deprotonating, or adding external cations/anions.
On these cases, protonation and consecutive CCS calculations can be done using:
weasel Ethiprole.sdf -W CCS-Prot-Ensemble
Deprotonation and consecutive CCS calculations can be done using:
weasel Ethiprole.sdf -W CCS-Deprot-Ensemble
If the input structure is already charged, a CCS calculation can be started by:
weasel Ethiprole.sdf -W CCS -c <charge>
Important
The -W CCS workflow conducts an optimization (GFN2-xTB) and a molecular charge calculation (B3LYP/def2-TZVP), but skips the protomer and conformer search.
Replace the <charge> with the total charge of the ion, e.g. 1 for a single positive charge, and -1 for a single negative charge.
Because of the flexible nature of Ethiprole, we used the CCS-Prot-Ensemble for this example.
When the calculation is finished, the final results are parsed into the .report and the .summary file.
The most relevant parts are shown in the following, but for more details,
see the decription of the output files.
The report file will look something like this, where the IDs of the structures, as well as their weights are reported:

Part of the report file printing the CCS values. The weights are the contributions of each structure to the total results calculated inside WEASEL by their energetical proportions.
In the summary file, only the most relevant informations are written:

Part of the summary file printing the CCS values.
If the calculation takes a while, don't worry! With 32 cores it should take about 1.5 hours, to give you a rough estimation. There are a lot of steps going on, so to understand how the final CCS values are computed, let's take a more detailed look at the individual workflow steps.
The steps of the workflow
The steps of the CCS-Prot-Ensemble and CCS-Deprot-Ensemble workflows are conducted as follows:
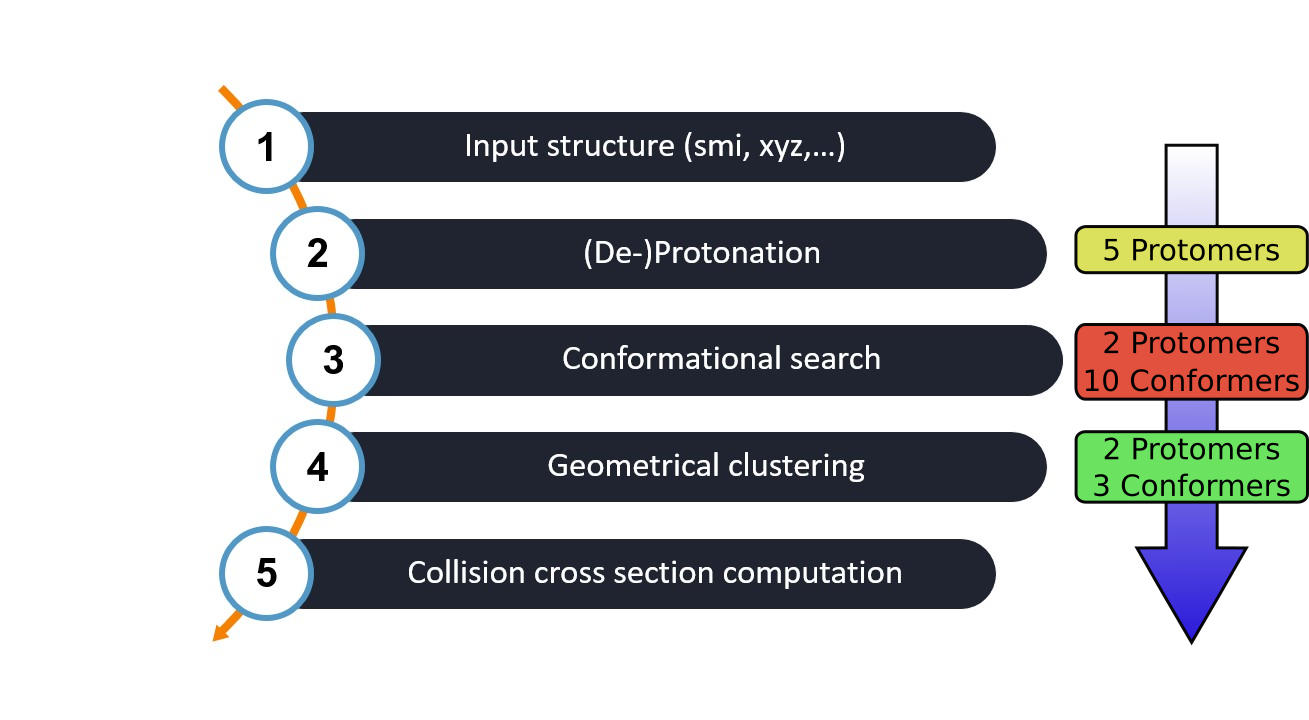
Overview of the CCS-(De-)Prot-Ensemble workflow steps inside WEASEL. The number of structures produced/reduced by each step for Ethiprole are added for reference.
Starting with an input structure (e.g. a SMILES string, XYZ file, etc.), the workflow follows a series of steps.
Protomer search of all possible (de-)protonation sites using the protomer search workflow.
Conformer search of all (de-)protonated structures with the conformer search tool. The preoptimization uses xtb at the GFN2-xTB level and filters structures with energies > 30 kcal/mol. Finally, clustering and single point calculations at the B3LYP/def2-TZVP level of theory filters out structures with an energy difference > 4 kcal/mol.
A special geometrical clustering of all conformers belonging to each protomer leads to the final structures, that are Boltzmann weighted for the last step.
Finally, the collision cross section calculations lead to the wanted results.
As you can see, the CCS calculation incorporates a lot of different steps of the WEASEL infrastructure. We will take a closer look at the specifics concerning the CCS files produced during the calculations and how to change the default behavior of the calculations.
The output files
A lot of files are produced during to the protomer search and the consecutive conformer search, that are described in detail on their corresponding help pages. Additionally, the CCS workflows will create a folder called CCS, in which the detailed output files are parsed. The folder structure is built up like the following:
.
├── Ethiprole.sdf
└── CCS
├── CCS_2
├── Ethiprole_Protomer_2_lowestProtomer_3_CCScalc.ccsc
├── Ethiprole_Protomer_2_lowestProtomer_3_CCScalc.out
└── Ethiprole_Protomer_2_lowestProtomer_3_CCScalc.xyz
└── Clusters
├── Ethiprole_CCS-Prot-Ensemble_CCScalc.clustering.out
├── Ethiprole_CCS-Prot-Ensemble_CCScalc.input.xyz
├── Protomer2_cluster1.xyz
└── Protomer2_cluster2.xyz
The CCS values are calculated inside WEASEL using the sub-program CCScalc, which directs its output into a folder named CCS_x. Here, the index x is a placeholder for the conformer ID of the structure that was calculated, e.g. conformer 49 can be found in folder CCS_49, conformer 2 in CCS_2, and so on. The folder contains the input and output files of the CCScalc program and looks like the following:
CCS Files |
Description |
|---|---|
Ethiprole_Protomer_2_lowestProtomer_3_CCScalc.ccsc |
input file specific to CCScalc |
Ethiprole_Protomer_2_lowestProtomer_3_CCScalc.out |
output file of CCScalc |
Ethiprole_Protomer_2_lowestProtomer_3_CCScalc.xyz |
structure input file in xyz format |
Inside the CCS main folder, a Clusters folder is created during the geometrical clustering in step 4 of the CCS workflows. Depending on the number of structures that are clustered, the number of clusters can vary. The files look like the following:
Clusters Files |
Description |
|---|---|
Ethiprole_CCS-Prot-Ensemble_CCScalc.clustering.out |
information on the clustering process |
Ethiprole_CCS-Prot-Ensemble_CCScalc.input.xyz |
input structures before clustering |
Protomer2_cluster1.xyz |
cluster 1 of protomer 2 |
Protomer2_cluster2.xyz |
cluster 2 of protomer 2 |
CCScalc
Usage
Inside WEASEL, the CCScalc program calculates the CCS of an input molecule by computing the mobility of an ion via molecular dynamics (MD) simulations based on Hamilton's equation of motion. For some more details, let's take a look at the CCScalc output file inside the CCS folder.

The CCScalc output of version 0.2. The most important parts are marked and commented.
The output file first lists the CCScalc settings that were used for the calculation, and then provides the CCS values and errors of each iteration of the calculations. Each CCS calculation is done a minimum number of times to obtain a more reliable result. Underneath, the summary provides the averaged CCS values, as well as the mobility value and the standard error of the mean.
Settings
The default settings can be manipulated with the following command-line arguments:
Warning
Changing the default values can drastically alter the results and if the values are lowered, the accuracy of the method will decrease!
Keyword |
Description |
Inside CCScalc |
Default |
|---|---|---|---|
|
number of velocity integration steps |
sets the accuracy for collision parameter b |
48 |
|
maximum number of cycles |
upper limit for convergence cycles |
40 |
|
minimum number of cycles |
lower limit for convergence cycles |
8 |
|
number of random stating geometries |
rotate molecule and vary distance Gas--Molecule |
768 |
|
the kind of collision gas |
set He or N2 |
N2 |
|
computation stopping criterion |
relative deviation of the standard error of the mean in percent |
0.35 |
|
switch on/off geometrical clustering |
--- |
on |
Warning
If the number of cycles is too small for the calculation to converge under the value provided by the -ccs-sem keyword, the program throws an error, but will still go through with the calculation.
Be advised: the results might not be considered statistically converged!
Note
If the number of maximum cycles is lower than the number of minimum cycles, the program will throw a warning but sets -ccs-mincycles to the value provided by the user (-ccs-maxcycles).
Note
CCScalc takes the temperature from the WEASEL input, which can be changed by setting -temp and the temperature in Kelvin via command line. The number of cores used for the calculation is the same as provided with WEASEL.
Note
The DFT charge calculation is important to obtain the correct di- and quadrupole contributions. The involved parametrization was conducted on the B3LYP/def2-TZVP level, which is taken as the standard level for the charge calculations. Changing this level could decrease the accuracy of the method!
Geometrical clustering
The larger the molecule, the more structures remain after step 3 in the CCS workflows (the conformer search), which is schematically depicted in the CCS workflow overview. While in the example of Ethiprole the number of conformers is small, larger molecules easily have hundreds of structures remaining after the conformational search.
However, computing the CCS value of all these structures is computationally demanding and having hundreds of CCS values can cause confusion concerning the "true" result.
Clustering the structures by their geometrical similarities thus reduces the number of structures that need to be calculated by CCScalc, decreasing
the timings significantly and provides a good overview on the relevant values.
When we take a look at the .results file of the calculation, we can see the geometrical clustering step looking like the following:

The geometrical clustering of the structures in step 4 of the CCS workflows.
Each protomer is clustered seperately and the number of protomers and conformers remaining after clustering is printed in the end. For these structures the CCS values are calculated and printed as seen above.